Cardiovascular diseases, as the leading cause of death worldwide, have their pathological basis, atherosclerosis, clearly identified as a chronic inflammatory disease. In this disease process, the immune system plays a central regulatory role, with innate immunity and adaptive immunity constituting a complex and sophisticated defense network[1]. In recent years, with the application of novel technologies such as single-cell sequencing and spatial transcriptomics, breakthrough progress has been made in our understanding of the mechanisms by which these two immune arms function in cardiovascular inflammation[2].
The initiation and progression of cardiovascular inflammation involve multiple stages, ranging from acute injury to chronic remodeling, each stage engaging distinct immune cell populations and molecular mechanisms. As the first line of defense, innate immunity responds rapidly to vascular injury within minutes to hours, recognizing pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) via pattern recognition receptors to initiate the inflammatory cascade[3]. In contrast, adaptive immunity requires days to weeks for full activation, establishing long-term immunological memory through the specific recognition and clonal expansion of T cells and B cells.
This review systematically elucidates the distinct functional characteristics of adaptive and innate immunity in cardiovascular inflammation, provides an in-depth analysis of the molecular mechanisms underlying innate immunity as the first line of defense against vascular injury, and explores the interactive network between these two immune arms in chronic vascular inflammation. Particular emphasis is placed on the key cytokines involved in adaptive immunity during post-infarction cardiac remodeling, followed by a critical assessment of the clinical implications of innate immune dysregulation in coronary artery disease.
Table of Contents
1. How do adaptive immunity and innate immunity differ in cardiovascular inflammation?
2. Why is innate immunity considered the first line of defense in vascular injury?
3. Interplay between innate and adaptive immunity in chronic vascular inflammation
4. Key mechanisms of adaptive immunity in regulating post-infarct cardiac remodeling
5. Clinical implications of dysregulated innate immunity in coronary artery disease
01 How do adaptive immunity and innate immunity differ in cardiovascular inflammation?
Innate and adaptive immune cells orchestrate indispensable roles in both the homeostasis maintenance and pathogenesis of the cardiovascular system. Elucidating the functional disparities between innate and adaptive immunity is crucial for deciphering the temporal progression of cardiovascular inflammation. The core distinctions between these two immune compartments are primarily manifested in three dimensions: initiation mechanisms, cellular composition, and stage-specific functional contributions to disease progression.
1.1 Initiation Mechanisms and Recognition Modes
In the initiation and progression of cardiovascular inflammation, innate and adaptive immunity exhibit fundamental differences in their response kinetics and recognition modalities.
Innate immunity (natural immunity) is characterized by its rapid activation, occurring within minutes to hours following vascular injury, a feature that establishes it as the first line of defense against vascular insult. Innate immunity relies on germline-encoded pattern recognition receptors (PRRs), including Toll-like receptor 2 (TLR2), TLR4, and nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3), to rapidly sense conserved pathogen-associated molecular patterns (PAMPs) and endogenous damage-associated molecular patterns (DAMPs), such as cholesterol crystals and oxidized low-density lipoprotein (oxLDL), in the absence of prior sensitization[4].
In contrast, the initiation of adaptive immunity entails a substantially prolonged timeframe, typically spanning several days to weeks. A defining feature of adaptive immunity is its exquisite antigenic specificity, conferred by T cell receptors (TCRs) and B cell receptors (BCRs) that recognize discrete antigenic epitopes. Generated via somatic recombination, these receptors display extraordinary structural diversity, enabling the recognition of a virtually unlimited repertoire of antigens. Within the context of cardiovascular inflammation, key autoantigens encompass oxLDL, heat shock proteins, and cardiac myosin.
1.2 Key Cell Populations and Functional Specialization
Within the microenvironment of cardiovascular inflammation, innate and adaptive immunity are composed of distinct cellular populations that exhibit both functional specialization and synergistic crosstalk.
The principal effector cells of innate immunity include monocytes/macrophages (innate lymphocytes), which internalize oxidized low-density lipoprotein (oxLDL) to form foam cells and display phenotypic plasticity between pro-inflammatory M1 and anti-inflammatory M2 polarization states, as well as neutrophils that release reactive oxygen species (ROS) and neutrophil extracellular traps (NETs) to participate in early debris clearance while contributing to tissue injury[5]. Additionally, this cell population encompasses dendritic cells (DCs) that act as the critical bridge linking innate and adaptive immunity through antigen presentation, natural killer (NK) cells, and the complement system.
The core cellular components of adaptive immunity encompass CD4+ T cells (including T helper 1 (Th1), Th2, Th17, and regulatory T cells (Tregs) ), CD8+ cytotoxic T cells, and B cells. Th1 cells amplify inflammatory responses via interferon-γ (IFN-γ) secretion, whereas Tregs constrain excessive inflammation through the production of interleukin-10 (IL-10) and transforming growth factor-β (TGF-β)[6]. B cells modulate immune responses either by generating pathogenic autoantibodies (e.g., anti-histone H2B antibodies) or by secreting regulatory cytokines[7].
1.3 Functional Characteristics at Different Stages of Vascular Inflammation
During the distinct developmental stages of cardiovascular inflammation, innate and adaptive immunity exert differential yet complementary roles. The progression of atherosclerosis is a continuous process spanning from early lesion formation to advanced complications, with each stage involving specific immune cell populations and molecular mechanisms.
In early atherogenesis (fatty streak phase), innate immunity plays a dominant role. Subendothelial low-density lipoprotein (LDL) undergoes oxidation, and the oxidized LDL triggers endothelial cells to upregulate adhesion molecules, including vascular cell adhesion molecule-1 (VCAM-1) and E-selectin, via Toll-like receptors, thereby recruiting monocytes and T cells. Infiltrating monocytes differentiate into macrophages via macrophage colony-stimulating factor (M-CSF) and internalize oxidized LDL through scavenger receptors, becoming foam cells. These cells secrete chemokines and cytokines, amplifying immune cell recruitment. Recent imaging studies reveal that cholesterol crystals appear in early lesions coincident with inflammatory cell influx. These crystals activate the NLRP3 inflammasome in macrophages, promoting the release of interleukin (IL)-1β and IL-18, that underscores the pivotal role of innate immunity in the initiation of atherosclerosis[7,8].
During the progressive phase of atherosclerosis (fibrous plaque stage), T cells in atherosclerosis, particularly CD4+ T cells, play an increasingly important role in adaptive immune responses. As lesions advance, increasing numbers of these T cells infiltrate the vascular wall and recognize autoantigens, such as oxidized low-density lipoprotein (oxLDL), presented by antigen-presenting cells. Activated T cells differentiate into distinct effector subsets, among which Th1 cells further activate macrophages through secretion of cytokines including IFN-γ, thereby establishing a positive inflammatory feedback loop. Additionally, T cells express CD40 ligand (CD40L), which engages CD40 on the surface of macrophages and B cells, potentiating the inflammatory response[8].
In the advanced stages of atherosclerosis (complex plaque stage), the interplay between innate and adaptive immunity becomes increasingly intricate. The necrotic core contains abundant cholesterol crystals, cellular debris, and inflammatory mediators, which persistently activate innate immune responses. Concurrently, adaptive immune responses peak during this phase, with T cells and B cells undergoing sustained activation within tertiary lymphoid organs formed in the adventitia, generating copious pro-inflammatory cytokines and autoantibodies.
During acute cardiovascular events such as myocardial infarction (MI), the rapid response of innate immunity again proves critical. Within hours after MI, substantial numbers of neutrophils and monocytes are recruited to the infarct site to clear necrotic tissue and initiate repair processes. While these early inflammatory responses are necessary for debridement and repair initiation, excessive or sustained inflammation may precipitate adverse cardiac remodeling and functional deterioration[8].
The post-infarction reparative phase is a complex process involving a precise transition from inflammation to repair. In this phase, adaptive immune cells, particularly regulatory T cells (Tregs), play crucial regulatory roles. Studies have shown that exogenous Treg infusion promotes the polarization of macrophages toward the anti-inflammatory M2 phenotype, reduces cardiomyocyte death, enhances angiogenesis, and improves cardiac function. These findings underscore the significant role of adaptive immunity in cardiovascular repair[8].
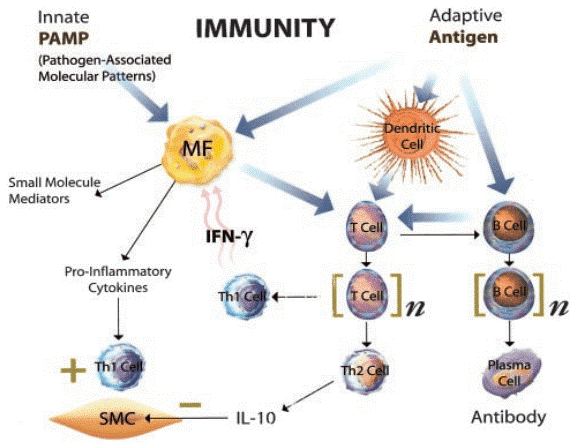
Fig. 1 Interplay between adaptive and innate immunity during atherogenesis[8].
02 Why is innate immunity considered the first line of defense in vascular injury?
As the “first responder” to vascular injury, innate immunity orchestrates inflammatory responses through precise molecular recognition and signal transduction. This section delineates the core molecular pathways mediating innate immune-driven vascular injury, focusing on PRR activation, inflammatory cell recruitment, and the dual roles of NETs and DAMPs.
2.1 Activation and Signal Transduction of Pattern Recognition Receptors (PRRs)
Upon ligand binding, toll-like receptor 4 (TLR4) activates the nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathways via both myeloid differentiation factor 88 (MyD88)-dependent and -independent mechanisms, thereby promoting the release of pro-inflammatory cytokines. The NLRP3 inflammasome, when activated by cholesterol crystals, mediates the caspase-1-dependent maturation and secretion of interleukin (IL)-1β and IL-18, which plays a pivotal role in early atherogenesis. As “conditional innate immune cells”, endothelial cells participate in inflammatory recruitment and antigen presentation through the activation of PRRs[9].
2.2 Inflammatory Cascade and Cellular Recruitment
Early-released cytokines, including interleukin (IL)-1β, tumor necrosis factor-α (TNF-α), and IL-6, synergize with chemokines such as monocyte chemoattractant protein-1 (MCP-1) and IL-8 to promote the recruitment of neutrophils and monocytes to injury sites via selectin- and integrin-mediated cascades of rolling, adhesion, and transendothelial migration. The complement system, when activated through classical and alternative pathways, generates the anaphylatoxins C3a and C5a and assembles the membrane attack complex (MAC), which directly damages vascular cells and amplifies the inflammatory response[8,9].
2.3 Dual Roles of Neutrophil Extracellular Traps (NETs) and Damage-Associated Molecular Patterns (DAMPs)
Neutrophil extracellular traps (NETs) ensnare pathogens via DNA-histone complexes, however, excessive NET release can induce endothelial injury and promote thrombogenesis. Damage-associated molecular patterns (DAMPs), including high-mobility group box 1 (HMGB1) and mitochondrial DNA, persistently activate pattern recognition receptors (PRRs), thereby establishing a deleterious “injury-inflammation feedback loop”, a phenomenon that is particularly pronounced in chronic kidney disease complicated by cardiovascular pathology[10].
03 Interplay between innate and adaptive immunity in chronic vascular inflammation
The chronicity and progression of cardiovascular inflammation are dependent on the dynamic crosstalk between innate and adaptive immunity. This section elaborates on how these two arms of immunity form a regulatory network and drive the persistent progression of the disease from three levels “initiation-feedback-positive feedback loops”.
3.1 Mechanisms by Which Innate Immunity Initiates Adaptive Immunity
The interaction between innate and adaptive immunity is a key process in the progression of cardiovascular inflammation, where innate immune cells, particularly dendritic cells (DCs), play an irreplaceable role in initiating adaptive immune responses. As professional antigen-presenting cells, DCs possess robust abilities to capture, process, and present antigens, serving as a critical bridge connecting innate and adaptive immunity.
In atherosclerotic lesions, DCs can capture various endogenous antigens, including oxidized low-density lipoprotein (oxLDL), heat shock proteins, and apoptotic cell debris. These antigens are processed into short peptide fragments within DCs, which then bind to major histocompatibility complex (MHC) class II molecules and are transported to the cell surface for recognition by CD4+ T cells. Recent studies have shown that DCs in atherosclerotic lesions not only localize to lesion sites but also migrate to local lymph nodes, where they present antigens to naive T cells to initiate adaptive immune responses[11].
The initiation of adaptive immunity by innate immunity also involves complex costimulatory signals. Studies indicate that DCs require “danger signals” to achieve full activation and acquire the capacity to activate T cells while capturing antigens. These danger signals, including pathogen-associated molecular patterns (PAMPs) from pathogens and damage-associated molecular patterns (DAMPs) from injured tissues, activate pattern recognition receptors (PRRs) such as toll-like receptors (TLRs) on DCs, thereby upregulating the expression of costimulatory molecules (e.g., B7-1/CD80, B7-2/CD86). Full T cell activation requires both antigen signals (via TCR-MHC complexes) and costimulatory signals (via CD28-B7 complexes); otherwise, T cells may enter anergic states or undergo apoptosis[12].
Recent studies have identified a specific B cell subset, innate response activator B cells (IRA B cells), that links innate and adaptive immunity. IRA B cells secrete granulocyte-macrophage colony-stimulating factor (GM-CSF) to promote the generation of conventional DCs, which further induce interferon-γ (IFN-γ)-producing Th1 cells. This fin9ding suggests that B cells not only act as effector cells in adaptive immunity but also regulate DC function to modulate the initiation of adaptive immunity[13].
Notably, the transition from innate to adaptive immunity also plays a critical role in immune responses following myocardial infarction (MI). Studies have demonstrated that innate immune cells (e.g., neutrophils and monocytes) predominantly infiltrate the infarcted myocardium in the early post-MI phase, followed by a gradual increase in T and B cell infiltration. This transition involves multiple mechanisms, including DAMPs released by injured myocardium activating DCs, which then migrate to draining lymph nodes to activate T cells[14].
3.2 Feedback Mechanisms of Adaptive Immunity in Regulating Innate Immunity
The regulation of innate immunity by adaptive immunity is a complex feedback process, primarily mediated by cytokines secreted by T cells and direct cell-cell interactions. This regulatory mechanism is critical for maintaining immune homeostasis, preventing excessive inflammation, and facilitating tissue repair.
T cell-derived cytokines are the principal mediators that modulate innate immune functions. Interferon-gamma (IFN-γ), a key cytokine secreted by T helper 1 (Th1) cells, potently enhances multiple macrophage functions, including phagocytosis, antigen-presenting capacity, and cytokine secretion. Within atherosclerotic lesions, IFN-γ promotes macrophage polarization toward the pro-inflammatory M1 phenotype, thereby amplifying their pro-inflammatory activities. Studies have demonstrated that IFN-γ additionally upregulates the expression of Toll-like receptors (TLRs) on macrophage surfaces, rendering these cells more sensitive to pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), and consequently establishing a positive inflammatory feedback loop[15].
Regulatory T cells (Tregs) play a unique anti-inflammatory role in regulating innate immunity. Tregs suppress the activation of innate immune cells through multiple mechanisms, including direct cell-contact inhibition, secretion of anti-inflammatory cytokines (e.g.,IL-10 and TGF-β), and local consumption of IL-2. Recent studies have shown that exogenously administered Tregs rapidly home to the infarcted myocardium following myocardial infarction (MI), acquire injury-specific transcriptomic profiles, and promote cardiac repair by modulating monocyte/macrophage functions. Specifically, Tregs facilitate macrophage polarization toward the anti-inflammatory M2 phenotype, reduce the secretion of pro-inflammatory cytokines, and concurrently enhance angiogenesis and collagen deposition[16].
CD4+ T cells also regulate splenic myeloid hematopoiesis following MI. Research indicates that CD4+ T cell-deficient mice exhibit significantly impaired splenic myeloid hematopoiesis after MI, whereas conventional CD4+ T cells promote myeloid hematopoiesis through both cell-contact-dependent and paracrine mechanisms, including IFN-γ signaling. Intriguingly, Treg depletion enhances in vivo myeloid hematopoiesis, as evidenced by increased numbers of progenitor cells and enhanced proliferative activity, suggesting that Tregs normally exert suppressive effects on myeloid hematopoiesis[15,16].
Antibodies secreted by B cells also modulate innate immune responses. Studies have demonstrated that natural antibodies, predominantly of the immunoglobulin M (IgM) isotype, recognize endogenous antigens (e.g., oxLDL) and promote their phagocytic clearance by macrophages through opsonization, thereby exerting protective effects. However, under certain conditions, antibodies may also exacerbate inflammatory responses. For example, anti-histone H2B antibodies accelerate atherosclerotic progression, likely via complement system activation or facilitation of immune complex formation[17].
3.3 Positive Feedback Loops Formed by Innate and Adaptive Immunity in Disease Progression
The positive feedback loops established between innate and adaptive immunity represent a critical mechanism driving the persistent progression and exacerbation of cardiovascular inflammation. These loops enable self-amplification of inflammatory responses, such that inflammation can persist and intensify even after the initial triggering stimuli have been eliminated.
In the pathogenesis and progression of atherosclerosis, a classic positive feedback loop involves reciprocal crosstalk between macrophages and T cells. As macrophages internalize oxLDL via scavenger receptors to form foam cells, they concomitantly secrete pro-inflammatory cytokines (e.g., IL-12), which promote T cell differentiation toward the Th1 phenotype. IFN-γ secreted by these Th1 cells, in turn, activates macrophages, enhancing their capacity for oxLDL uptake and pro-inflammatory cytokine secretion, thereby establishing a “macrophage-T cell” positive feedback loop[18].
Another important positive feedback loop involves interactions between the complement system and inflammatory cells. The complement activation products C3a and C5a recruit and activate inflammatory cells (e.g., neutrophils and monocytes), which subsequently release inflammatory mediators that further amplify complement activation. Following acute myocardial infarction (MI), complement activation has been shown to induce endothelial glycocalyx degradation and endothelial dysfunction via the C5a/C5a receptor 1 (C5aR1) axis, and this endothelial dysfunction further promotes inflammatory cell infiltration and complement activation[19].
Neutrophil extracellular trap (NET) formation also contributes to the establishment of positive feedback circuitry. NETs generated by neutrophils not only directly injure vascular endothelial cells but also act as damage-associated molecular patterns (DAMPs) to activate other innate immune cells, thereby promoting the release of inflammatory mediators. Studies have demonstrated that histones within NETs activate platelets and the coagulation system, facilitating thrombogenesis. Thrombosis, in turn, induces tissue ischemia and injury, leading to the release of additional DAMPs that further activate innate immunity[20].
Recent studies have further elucidated a positive feedback loop involving IL-1β and the NLRP3 inflammasome. Beyond serving as a product of NLRP3 inflammasome activation, IL-1β induces cellular expression of additional NLRP3 and pro-IL-1β, thereby forming a self-amplifying circuit. Within atherosclerotic lesions, cholesterol crystals activate the NLRP3 inflammasome to produce IL-1β, this IL-1β, in turn, promotes further cholesterol crystallization and inflammatory cell infiltration, exacerbating lesion progression[21].
The existence of these positive feedback loops endows cardiovascular inflammation with its characteristic chronic and progressive properties. Even after the elimination of inciting factors (e.g., via lipid-lowering interventions), inflammatory responses may persist and continue to progress. Consequently, disrupting these positive feedback loops has emerged as a pivotal therapeutic strategy for cardiovascular diseases, including approaches such as anti-inflammatory agents, immunomodulatory interventions, and complement cascade inhibition.
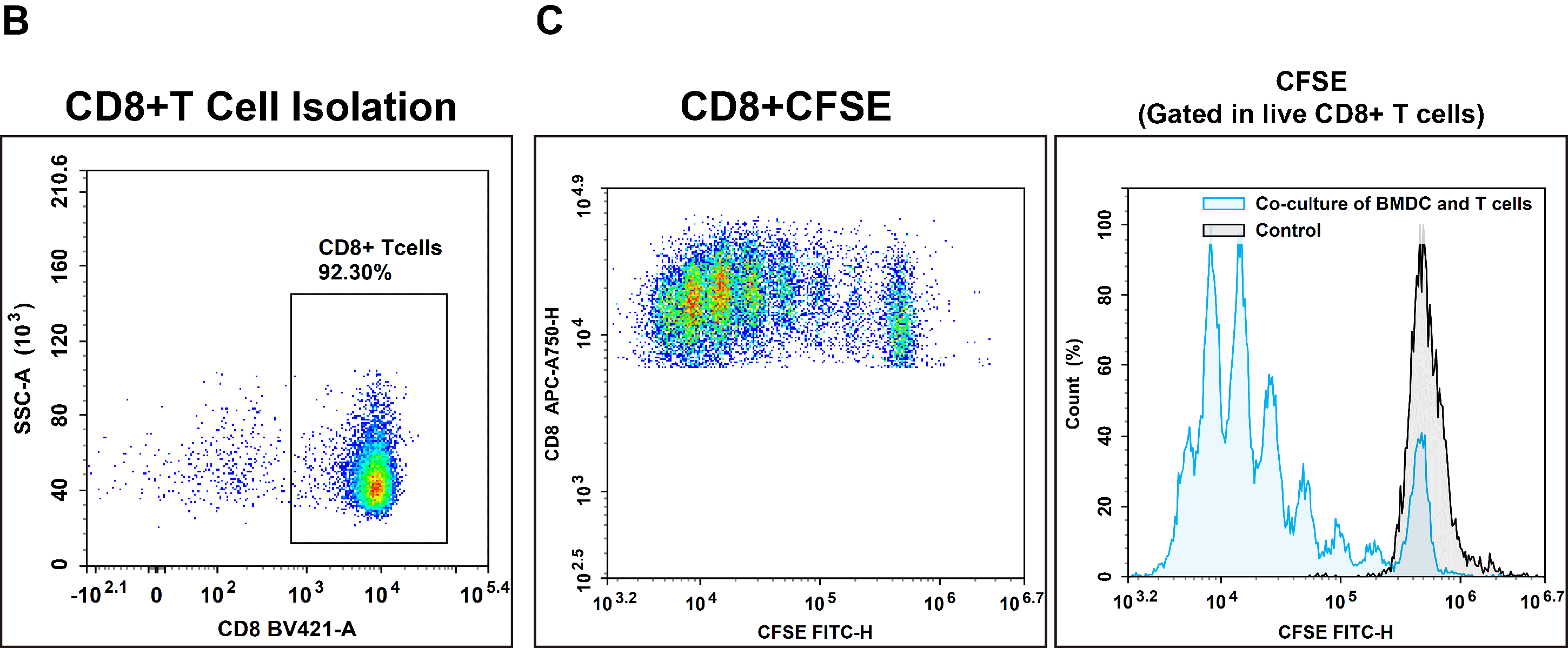
Fig. 2 Interaction between T cells and bone marrow-derived dendritic cells (BMDCs). CD8+ T cells were isolated from the splenocytes of C57BL/6 mice using EasySort™ Mouse CD8+T Cell Isolation Kit (cat. no. MIM003N). The purity of the isolated CD8⁺ T cells was verified by flow cytometry. Following CFSE labeling, CD8⁺ T cells were co‑cultured with mature BMDCs for 72 h in vitro, and the proliferation of CD8⁺ T cells was subsequently assessed. (The datas are provided by Elabceience.)
Elabscience® Quick Overview of Popular Products:
Table 1. Reagents for Macrophages Research
|
Product Name |
Cat. No. |
|
EasySort™ Mouse CD8+T Cell Isolation Kit |
MIM003N |
|
EasySort™ Mouse CD3+T Cell Isolation Kit |
MIM001N |
|
EasySort™ Mouse CD4+T Cell Isolation Kit |
MIM002N |
|
EasySort™ Human CD3+T Cell Isolation Kit |
MIH001N |
|
Mouse Bone Marrow-derived Dendritic Cells (BMDC) Induction and Identification Kit |
XJM003 |
|
APC Anti-Mouse CD8 Antibody[YTS-169] |
AN00576E |
|
Elab Fluor® Violet 450 Anti-Mouse CD8a Antibody[53-6.7] |
E-AB-F1104Q |
|
Caspase 1 Activity Assay Kit(Colorimetric Method) |
E-CK-A381 |
|
Caspase 1 Activity Detection Substrate for Flow Cytometry |
E-CK-A481 |
|
Reactive Oxygen Species (ROS) Fluorometric Assay Kit (Red) |
E-BC-F005 |
|
Lactate Dehydrogenase (LDH) Cytotoxicity Colorimetric Assay Kit |
E-BC-K771-M |
|
FITC Anti-Human IL-1 beta Antibody[CRM56] |
AN00842C |
|
Human IL-1β(Interleukin 1 Beta) ELISA Kit |
E-EL-H0149 |
|
Mouse IL-1β(Interleukin 1 Beta) ELISA Kit |
E-EL-M0037 |
|
Rat IL-1β(Interleukin 1 Beta) ELISA Kit |
E-EL-R0012 |
04 Key mechanisms of adaptive immunity in regulating post-infarct cardiac remodeling
Cardiac remodeling following myocardial infarction results from an imbalance between inflammation and repair, a process in which adaptive immunity plays a central regulatory role. This section elucidates the key mechanisms by which adaptive immunity influences post-infarct remodeling, focusing on cytokine balance, immune cell dynamics, and immune checkpoint regulation.
4.1 Cytokine Network Balance
The dynamic changes in immune cells subsequent to myocardial infarction constitute a highly ordered process, which is characterized by the temporally orchestrated recruitment, activation, and functional transitions of various immune cell subsets. This intricate process is tightly regulated by a complex cytokine network, whose dysregulation or balance ultimately dictates the outcome of cardiac remodeling. Specifically, Th1-type cytokines, including interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α), tend to exacerbate myocardial injury, whereas Th2-type cytokines such as interleukin-4 (IL-4) and interleukin-13 (IL-13) facilitate M2 macrophage polarization and promote tissue repair. Moreover, regulatory T cells (Tregs) modulate macrophage phenotypic switching and attenuate cardiac fibrosis through the secretion of interleukin-10 (IL-10) and Nestin-1. In addition, interleukin-17A (IL-17A), which is predominantly derived from γδ T cells, mediates cardiac fibrosis via an interleukin-1β (IL-1β)-dependent mechanism, and its circulating levels are closely correlated with adverse clinical prognosis[22].
4.2 Dynamic Remodeling of Immune Cell Populations
Within 24 hours post-myocardial infarction, the proportion of monocytes markedly increases to 62.4%, as opposed to 30.4% in controls, whereas T cell frequencies exhibit a concurrent decline. Between days 7 and 14 following infarction, infiltration by adaptive immune cells gradually escalates, with regulatory T cells (Tregs) homing to the infarcted myocardium and adopting injury-specific phenotypic signatures. Through these adaptations, Tregs modulate splenic myelopoiesis and contribute to the resolution of local inflammation[23].
4.3 Regulatory Role of Immune Checkpoint Molecules
The programmed cell death protein 1/programmed death-ligand 1 (PD-1/PD-L1) axis functions as a critical negative regulatory checkpoint that curbs excessive inflammatory responses. Nevertheless, therapeutic blockade of this pathway has been linked to an elevated risk of precipitating myocarditis. Conversely, V-set and immunoglobulin domain-containing 4 (VSIG4) aggravates post-infarction inflammation by dampening the anti-inflammatory functions of CD16+ monocytes, thereby positioning itself as a promising therapeutic target for mitigating adverse cardiac remodeling[24].
05 Clinical implications of dysregulated innate immunity in coronary artery disease
Advances in our understanding of the role of innate immunity in coronary artery disease (CAD) have fueled the rapid development of targeted therapeutic strategies, which have translated into novel prospects for the prevention and clinical management of this prevalent cardiovascular disorder.
Upon activation of innate immunity, the rapid release of inflammatory cytokines constitutes the core of the early vascular inflammatory response to injury. These mediators not only act directly on vascular cells but also facilitate the recruitment and activation of additional immune cells, thereby propagating a robust inflammatory cascade.
Among these cytokines, interleukin-1β (IL-1β) stands out as a pivotal proinflammatory cytokine generated following innate immune activation. In the context of cardiovascular inflammation, IL-1β is primarily secreted by activated macrophages and monocytes via a tightly regulated two-step mechanism. This process involves the transcriptional upregulation of pro-IL-1β, which is triggered by pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs), followed by the cleavage of pro-IL-1β into its biologically active mature form by caspase-1, which is a protease activated downstream of the NLRP3 inflammasome. Accumulating evidence indicates that the expression level of IL‑1β is closely correlated with the severity of atherosclerotic lesions. Consistently, pharmacological inhibition or genetic ablation of the IL-1β signaling pathway significantly attenuates both the initiation and progression of atherosclerosis[25].
Numerous clinical studies have demonstrated that canakinumab, a specific IL-1β inhibitor, significantly reduces the risk of major adverse cardiovascular events (MACE) in patients post-myocardial infarction (post-MI), with a hazard ratio (HR) of 0.85. This finding thus validates the therapeutic potential of the NLRP3-IL-1β signaling axis in cardiovascular diseases[25].
In parallel with direct IL-1β inhibition, considerable efforts have been devoted to the development of inhibitors targeting the NLRP3 inflammasome. Among these agents, MCC950, a novel NLRP3-specific inhibitor, has exhibited favorable safety and efficacy profiles in preclinical studies. This agent exerts its anti-inflammatory effects by suppressing the assembly and activation of the NLRP3 inflammasome, thereby reducing the production of IL-1β and IL-18, which are two key pro-inflammatory cytokines[26].
As discussed earlier, therapeutic strategies targeting the complement system have also shown considerable promise in the management of CAD. Given the well-established role of complement activation in both the pathogenesis of atherosclerosis and the progression of acute coronary syndromes (ACS), pharmacological inhibition of this pathway may represent a viable therapeutic approach. For instance, eculizumab, a monoclonal antibody targeting complement component C5, has demonstrated substantial efficacy in the treatment of diseases such as paroxysmal nocturnal hemoglobinuria (PNH), and its potential application in cardiovascular diseases is currently under active investigation[27].
Another emerging area of research involves therapeutic strategies targeting neutrophil extracellular trap (NET) formation. Accumulating evidence indicates that NETs play a pivotal role in the pathogenesis of ACS and thrombotic events, suggesting that inhibition of NET formation may contribute to the prevention of adverse cardiovascular events. Potential approaches in this regard include the administration of DNase I to degrade the DNA scaffold of NETs, as well as pharmacological inhibition of peptidylarginine deiminase 4 (PADI4) to block histone citrullination, which is a key regulatory step in NETosis[28].
In addition, in the context of post-MI management, strategies aimed at modulating the innate immune response are also being actively explored. Mounting evidence suggests that early administration of anti-inflammatory agents following MI may reduce infarct size and improve long-term clinical outcomes. For example, administration of colchicine within 24 hours of MI onset has been shown to significantly reduce the risk of the composite endpoint, including cardiovascular death, recurrent MI, or stroke[29].
Complementing these therapeutic approaches, strategies targeting damage-associated molecular patterns (DAMPs) are also under active investigation. As previously noted, elevated levels of multiple DAMPs, which include heat shock protein 70 (Hsp70), hyaluronan, high-mobility group box 1 (HMGB1), and calprotectin, have been documented in patients with chronic kidney disease (CKD). Neutralizing antibodies or receptor antagonists directed against these DAMPs may therefore help attenuate vascular inflammation and improve cardiovascular outcomes in this high-risk population[30].
Furthermore, precision therapeutic approaches guided by inflammatory phenotypes, such as NLRP3-driven or complement-predominant subtypes, may represent the future trajectory of cardiovascular intervention. However, several challenges must be addressed, including the standardization of biomarkers, assessment of long-term therapeutic safety (particularly infectious risk), and considerations of treatment accessibility and affordability[31].
In conclusion, cardiovascular immunology, as a rapidly evolving interdisciplinary field, is profoundly reshaping our understanding of and therapeutic strategies for cardiovascular diseases. With advancing research and technological progress, there is compelling reason to believe that immunomodulatory therapies will ultimately translate into improved prognosis and enhanced quality of life for patients with cardiovascular disease.
References:
[1] Hansson G K, Libby P, Schönbeck U, et al. Innate and adaptive immunity in the pathogenesis of atherosclerosis[J]. Circulation research, 2002, 91(4): 281-291.
[2] Cetin E, Raby A C. Understanding atherosclerotic plaque cellular composition: recent advances driven by single cell omics[J]. Cells, 2025, 14(11): 770.
[3] Matter M A, Paneni F, Libby P, et al. Inflammation in acute myocardial infarction: the good, the bad and the ugly[J]. European heart journal, 2024, 45(2): 89-103.
[4] Zhang B, Liu Y, Sui Y B, et al. Cortistatin inhibits NLRP3 inflammasome activation of cardiac fibroblasts during sepsis[J]. Journal of cardiac failure, 2015, 21(5): 426-433.
[5] Krishnan J, Hennen E M, Ao M, et al. NETosis drives blood pressure elevation and vascular dysfunction in hypertension[J]. Circulation Research, 2024, 134(11): 1483-1494.
[6] Hofmann U, Frantz S. Role of T-cells in myocardial infarction[J]. European heart journal, 2016, 37(11): 873-879.
[7] Bird G, Hally K, La Flamme A, et al. The Role of B Cells in Acute Myocardial Infarction[J]. Heart, Lung and Circulation, 2019, 28: S18.
[8] Hansson G K, Libby P, Schönbeck U, et al. Innate and adaptive immunity in the pathogenesis of atherosclerosis[J]. Circulation research, 2002, 91(4): 281-291.
[9] Faure E, Equils O, Sieling P A, et al. Bacterial lipopolysaccharide activates NF-κB through Toll-like receptor 4 (TLR-4) in cultured human dermal endothelial cells: differential expression of TLR-4 and TLR-2 in endothelial cells[J]. Journal of Biological Chemistry, 2000, 275(15): 11058-11063.
[10] Cicco S, Cicco G, Racanelli V, et al. Neutrophil extracellular traps (NETs) and damage‐associated molecular patterns (DAMPs): two potential targets for COVID‐19 treatment[J]. Mediators of inflammation, 2020, 2020(1): 7527953.
[11] Nickel T, Schmauss D, Hanssen H, et al. oxLDL uptake by dendritic cells induces upregulation of scavenger-receptors, maturation and differentiation[J]. Atherosclerosis, 2009, 205(2): 442-450.
[12] Habets K L L, Van Puijvelde G H M, Van Duivenvoorde L M, et al. Vaccination using oxidized low-density lipoprotein-pulsed dendritic cells reduces atherosclerosis in LDL receptor-deficient mice[J]. Cardiovascular research, 2010, 85(3): 622-630.
[13] Hilgendorf I, Theurl I, Gerhardt L M S, et al. Innate response activator B cells aggravate atherosclerosis by stimulating T helper-1 adaptive immunity[J]. Circulation, 2014, 129(16): 1677-1687.
[14] Hofmann U, Frantz S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction[J]. Circulation research, 2015, 116(2): 354-367.
[15] Li C, Zong W, Zhang M, et al. Increased ratio of circulating T-Helper 1 to T-Helper 2 cells and severity of coronary artery disease in patients with acute myocardial infarction: a prospective observational study[J]. Medical science monitor: international medical journal of experimental and clinical research, 2019, 25: 6034.
[16] Xia N, Lu Y, Gu M, et al. A unique population of regulatory T cells in heart potentiates cardiac protection from myocardial infarction[J]. Circulation, 2020, 142(20): 1956-1973.
[17] Itakura M, Yamaguchi K, Kitazawa R, et al. Histone functions as a cell-surface receptor for AGEs[J]. Nature Communications, 2022, 13(1): 2974.
[18] Ma P, Liu J, Qin J, et al. Expansion of pathogenic cardiac macrophages in immune checkpoint inhibitor myocarditis[J]. Circulation, 2024, 149(1): 48-66.
[19] Frangogiannis N G. The inflammatory response in myocardial injury, repair, and remodelling[J]. Nature Reviews Cardiology, 2014, 11(5): 255-265.
[20] Döring Y, Libby P, Soehnlein O. Neutrophil extracellular traps participate in cardiovascular diseases: recent experimental and clinical insights[J]. Circulation research, 2020, 126(9): 1228-1241.
[21] Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction[J]. Nature Reviews Cardiology, 2018, 15(4): 203-214.
[22] Yuan D, Tie J, Xu Z, et al. Dynamic Profile of CD4+ T‐Cell‐Associated Cytokines/Chemokines following Murine Myocardial Infarction/Reperfusion[J]. Mediators of Inflammation, 2019, 2019(1): 9483647.
[23] van Blokland I V, Oelen R, Groot H E, et al. Single-cell dissection of the immune response after acute myocardial infarction[J]. Circulation: Genomic and Precision Medicine, 2024, 17(3): e004374.
[24] Erbe S, Kneuer J, Buettner P, et al. VSIG4 inhibits the anti-inflammatory reaction of immune cells after cardiogenic shock[J]. European Heart Journal, 2024, 45(Supplement_1): ehae666. 3810.
[25] Everett B M, Cornel J H, Lainscak M, et al. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure[J]. Circulation, 2019, 139(10): 1289-1299.
[26] Coll R C, Schroder K, Pelegrín P. NLRP3 and pyroptosis blockers for treating inflammatory diseases[J]. Trends in pharmacological sciences, 2022, 43(8): 653-668.
[27] Ramoni D, Carbone F, Kraler S, et al. Inflammation-driven plaque erosion in atherosclerosis: a focus on complement system pathways[J]. Current Atherosclerosis Reports, 2025, 27(1): 42.
[28] Döring Y, Soehnlein O, Weber C. Neutrophils cast NETs in atherosclerosis: employing peptidylarginine deiminase as a therapeutic target[J]. Circulation Research, 2014, 114(6): 931-934.
[29] Mewton N, Roubille F, Bresson D, et al. Effect of colchicine on myocardial injury in acute myocardial infarction[J]. Circulation, 2021, 144(11): 859-869.
[30] Mazzarino M, Cetin E, Bartosova M, et al. Therapeutic targeting of chronic kidney disease-associated DAMPs differentially contributing to vascular pathology[J]. Frontiers in immunology, 2023, 14: 1240679.
[31] Everett B M, Cornel J H, Lainscak M, et al. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure[J]. Circulation, 2019, 139(10): 1289-1299.

