Ischemic heart disease (IHD) remains a leading cause of morbidity and mortality globally, primarily attributed to the irreversible loss of cardiomyocytes subsequent to myocardial infarction (MI) and the consequent pathological remodeling of the ventricular wall. The myocardial infarction inflammation, a post-ischemic inflammatory response, is the critical determinant of both the immediate magnitude of tissue damage and the long-term progression toward heart failure. Although essential for clearing necrotic debris and initiating tissue repair, an exaggerated or dysregulated inflammatory cascade exacerbates cardiomyocyte injury and promotes adverse fibrosis. Among the numerous immune cells recruited to the ischemic myocardium, innate immune effectors, particularly macrophages and immune system natural killer cells, have emerged as central coordinators of this inflammatory process. Macrophages play a pivotal role in phagocytosing dead cells and orchestrating tissue repair; however, their pro-inflammatory activation can amplify tissue injury. Meanwhile, natural killer (NK) cells, traditionally recognized for their roles in antiviral and antitumor immunity, are now known to significantly modulate the inflammatory microenvironment in IHD through direct cytotoxic effects and the secretion of potent cytokines[1].
This article reviews the pivotal role of macrophages in driving inflammatory responses following myocardial infarction, the mechanisms by which NK cells exacerbate tissue damage through inflammatory pathways in ischemic heart disease, the specific cytokine profiles secreted by both macrophages and NK cells during ischemic injury, and how NK cell-derived interferon gamma (IFN-γ) critically modulates macrophage activation, thereby shaping the inflammatory microenvironment in ischemic heart disease.
Table of Contents
1. Macrophages driving inflammatory responses after myocardial infarction
2. NK cells promote tissue damage through inflammatory pathways in ischemic heart disease
3. Cytokine secretion by macrophages and nk cells in ischemic heart disease
4. NK cell-derived IFN-γ regulates macrophage activation in ischemic heart disease
01 Macrophages driving inflammatory responses after myocardial infarction
Macrophages play a critical role in orchestrating inflammatory responses in the aftermath of MI. Immediately following an MI, necrotic cardiomyocytes release damage-associated molecular patterns (DAMPs), which act as potent danger signals to initiate innate immune activation. Resident cardiac macrophages are rapidly stimulated by these DAMPs, while circulating monocytes are recruited to the infarct region and differentiate into pro-inflammatory macrophages, typically categorized as M1-like cells[2, 3]. These early phase macrophages are indispensable for the initial debridement process, efficiently phagocytosing dead cells and cellular debris. However, this activity is accompanied by the secretion of a robust cocktail of pro-inflammatory mediators, including reactive oxygen species (ROS), interleukin 1β (IL-1β), interleukin 6 (IL-6), and tumor necrosis factor α (TNF-α)[4]. While this inflammatory surge is essential for wound clearance, its excessive activation or prolonged duration inflicts substantial collateral damage on the adjacent viable myocardium. This not only exacerbates the initial ischemic injury but also drives adverse ventricular remodeling[5], making the balance between necessary debridement and destructive inflammation a key determinant of macrophages role in inflammation and post MI clinical outcomes.
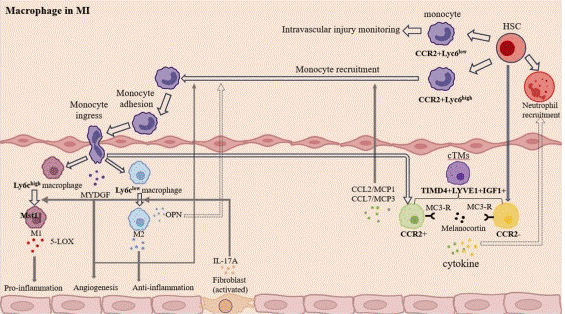
Fig. 1 The role of macrophages in MI is summarized. In mouse, a majority of Ly6chigh monocytes and macrophages release pro-inflammatory cytokines and break down infarcted tissue and cell debris in the early stages of a myocardial infarction. This phase is followed by Ly6clow macrophages that are anti-inflammatory and aid in the reduction of inflammation and the healing of wounds[3].
02 NK cells promote tissue damage through inflammatory pathways in ischemic heart disease
NK cells exacerbate tissue injury in IHD through NK cell cytotoxicity and by driving pro-inflammatory pathways. As one of the first lymphocyte subsets to infiltrate the infarcted myocardium, NK cells arrive as early as several hours to days post MI[6]. Their primary pathogenic mechanism is NK cell cytotoxicity, which relies on NK cell activation: stressed yet viable cardiomyocytes and endothelial cells in the peri-infarct region upregulate ligands for NKG2D, a key activating receptor on NK cells[7]. This receptor- ligand interaction triggers the release of perforin and granzyme B from NK cells, leading to the lysis of these stressed host cells and thus expanding the injury beyond the initial ischemic core. This secondary wave of cell death not only increases the total infarct size but also releases additional DAMPs. These DAMPs further amplify the inflammatory cascade by activating macrophages and other immune cells, creating a vicious cycle of inflammation and tissue destruction that impairs cardiac function and drives maladaptive reparative processes[1].
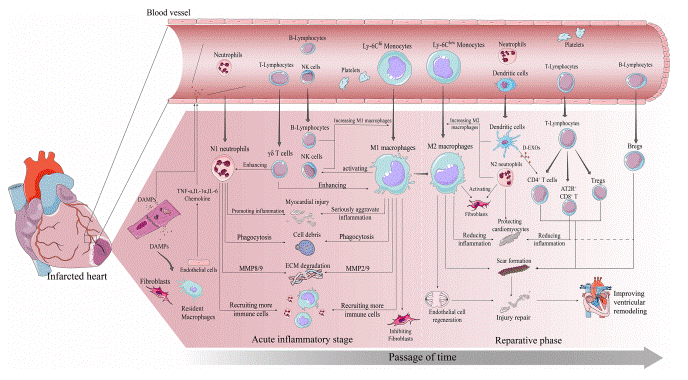
Fig. 2 Immune cascade after MI. Early post-MI necrotic cells release DAMPs which bind to surviving parenchymal cells releasing pro-inflammatory and chemokines and recruiting immune cells to infiltrate[6].
03 Cytokine secretion by macrophages and nk cells in ischemic heart disease
In IHD, macrophages and NK cells secrete distinct cytokines that orchestrate the inflammatory immune response. Although their functional roles partially overlap, their cytokine secretion profiles are uniquely distinct and mutually complementary, thereby establishing a synergistic inflammatory microenvironment. Macrophages serve as prominent producers of IL 1β, TNF α, IL 6, and the chemokine CCL2 (MCP 1), which are critical for recruiting additional monocytes and other leukocytes to the injured myocardial tissue[8]. By contrast, NK cells represent a major early cellular source of IFN γ, together with TNF α and granulocyte macrophage colony stimulating factor (GM CSF), a key macrophage activating factor[9]. IFN γ acts as a master regulator of ifn gamma macrophage activation and polarization, whereas GM CSF supports the differentiation and survival of inflammatory myeloid lineage cells. The concerted actions of these cytokines, including macrophage derived IL 1β and TNF α that directly induce cardiomyocyte dysfunction and extracellular matrix degradation, together with NK cell derived IFN γ that shapes macrophage polarization, collectively establish a robust inflammatory milieu[10]. When dysregulated, this milieu drives the progression of IHD from acute myocardial injury toward chronic heart failure[11].
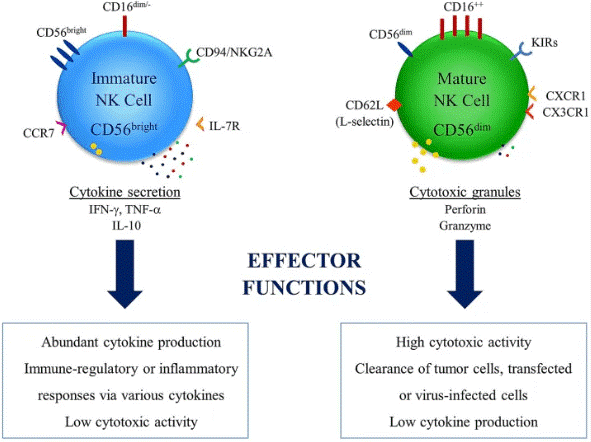
Fig. 3 Differential effector functions of NK cell subsets. NK cells have two major subsets in relation with their CD56 expression levels. High expression levels of CD56 is observed in immature NK cell subset[9].
04 NK cell-derived IFN-γ regulates macrophage activation in ischemic heart disease
NK cell-derived IFN-γ modulates macrophage activation in ischemic heart disease, thereby representing a critical node of crosstalk between these two innate immune cell populations[12]. As a signature cytokine of NK cells and T helper 1 (Th1) cells, IFN-γ serves as a potent polarizing stimulus for macrophages[13], and its production can be quantified using an IFN-γ assay (ifn gamma assay). Upon binding to the IFN-γ receptor on macrophages, IFN-γ triggers the JAK–STAT1 signaling cascade, which transcriptionally reprograms macrophages toward a classical pro-inflammatory M1 phenotype. This polarization enhances the capacity of macrophages to secrete elevated levels of inflammatory cytokines such as IL-12 and TNF-α, increases the expression of major histocompatibility complex class II (MHC-II) molecules for efficient antigen presentation, and upregulates inducible nitric oxide synthase (iNOS), thereby promoting nitric oxide (NO) production[14]. This positive feedback loop, whereby NK cell-derived IFN-γ licenses macrophages to mount an augmented inflammatory response, constitutes a key mechanism that sustains and amplifies inflammation during the early post-myocardial infarction (post-MI) period. While essential for host defense against pathogens, this crosstalk becomes detrimental in the sterile inflammatory milieu of MI, as it delays the critical phenotypic transition of macrophages from a pro-inflammatory M1 state toward a reparative M2 state, which is indispensable for inflammation resolution and effective scar formation[1]. Therapeutic strategies targeting this specific NK cell, macrophage axis, such as IFN-γ neutralization, therefore represent a promising approach to attenuate excessive inflammation and enhance cardiac repair following MI[1].
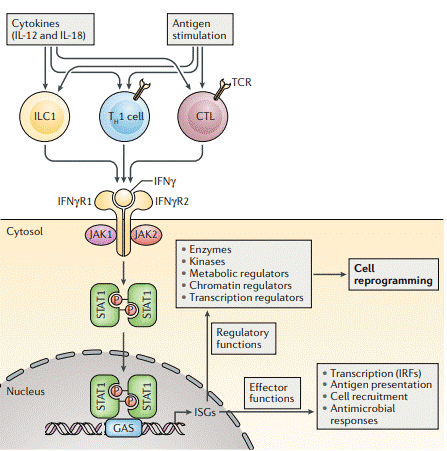
Fig. 4 IFN-γ production and signalling. IFN-γ is produced by innate-like lymphocytes, including group 1 innate lymphoid cells (ILC1s), and by adaptive lymphocytes, including TH1 cells and cytotoxic T lymphocytes (CTLs), in response to cytokine and antigen stimulation. IFN-γ acts on its receptor to induce rapid and transient Janus kinase (JAK) -signal transducer and activator of transcription (STAT) signalling and interferon-stimulated gene (ISG) induction. Over time, the cellular IFN-γ response evolves by impacting the expression and function of various enzymes and regulators of metabolism, chromatin and transcription to induce a reprogrammed cellular state that is characterized not only by its gene expression profile but also by altered responsiveness to environmental challenges[13].
In summary, macrophages and NK cells play central roles in the inflammatory response of ischemic heart disease: macrophages are recruited and activated by DAMPs post-myocardial infarction to clear debris but secrete pro-inflammatory mediators that can exacerbate tissue damage. NK cells infiltrate early and exert NK cell cytotoxicity while secreting cytokines like IFN-γ, and NK cell-derived IFN-γ modulates macrophage polarization toward a pro-inflammatory phenotype, together shaping the inflammatory microenvironment that influences cardiac repair and disease progression.
Elabscience® Quick Overview of Popular Products:
Table 1. Research Tools for IHD
|
Cat. No. |
Product Name |
|
E-BC-K138-F |
Reactive Oxygen Species (ROS) Fluorometric Assay Kit (Green) |
|
E-BC-K135-M |
Nitric Oxide (NO) Colorimetric Assay Kit (Nitrate Reductase Method) |
|
E-HSEL-H0003 |
High Sensitivity Human IL-6 (Interleukin 6) ELISA Kit |
|
E-OSEL-H0001 |
QuicKey Pro Human IL-6(Interleukin 6) ELISA Kit |
|
CQH001 |
CellaQuant™ Human IL-6 (Interleukin 6) ELISA Kit |
|
ESP-H0009S |
Human IL-6 (Interleukin 6) solid ELISPOT Kit |
|
E-EL-H1617 |
Human GzmB(Granzyme B) ELISA Kit |
|
E-HSEL-H0001 |
High Sensitivity Human IL-1β (Interleukin 1 Beta) ELISA Kit |
|
E-EL-H0109 |
Human TNF-α(Tumor Necrosis Factor Alpha) ELISA Kit |
|
E-HSEL-H0007 |
High Sensitivity Human IFN-γ (Interferon Gamma) ELISA Kit |
|
MIM005N |
EasySort™ Mouse NK Cell Isolation Kit |
|
E-EL-H0081 |
Human GM-CSF ELISA Kit |
|
E-EL-H0150 |
Human IL-12(Interleukin 12) ELISA Kit |
|
E-EL-H0753 |
Human NOS2/iNOS(Nitric Oxide Synthase 2, Inducible) ELISA Kit |
References:
[1] Kumrić, M., et al., The Role of Natural Killer (NK) Cells in Acute Coronary Syndrome: A Comprehensive Review. Biomolecules, 2020. 10(11): p. 1514.
[2] Zhao, Y., et al., Comprehensive macro and micro views on immune cells in ischemic heart disease. Cell Proliferation, 2024. 57(12): p. e13725.
[3] Zhuang, Q., et al., Recent advances in potential targets for myocardial ischemia reperfusion injury: Role of macrophages. Molecular Immunology, 2024. 169: p. 1-9.
[4] Xu, A., et al., Dynamic Regulation of Macrophage Polarization in Acute Myocardial Infarction and Its Therapeutic Potential. Journal of Inflammation Research, 2025. 18(null): p. 17363-17385.
[5] Chen, R., et al., Macrophages in cardiovascular diseases: molecular mechanisms and therapeutic targets. Signal Transduction and Targeted Therapy, 2024. 9(1): p. 130.
[6] Nian, W., Z. Huang, and C. Fu, Immune cells drive new immunomodulatory therapies for myocardial infarction: From basic to clinical translation. Frontiers in Immunology, 2023.14:1097295.
[7] Shao, L., et al., Inflammation in myocardial infarction: roles of mesenchymal stem cells and their secretome. Cell Death Discovery, 2022. 8(1): p. 452.
[8] Dewald, O., et al., CCL2/Monocyte Chemoattractant Protein-1 Regulates Inflammatory Responses Critical to Healing Myocardial Infarcts. Circulation Research, 2005. 96(8): p. 881-889.
[9] Kucuksezer, U.C., et al., The Role of Natural Killer Cells in Autoimmune Diseases. Frontiers in Immunology, 2021.12:622306.
[10] Ong, S., N.R. Rose, and D. Čiháková, Natural killer cells in inflammatory heart disease. Clinical immunology, 2017. 175: p. 26-33.
[11] Nian, M., et al., Inflammatory cytokines and postmyocardial infarction remodeling. Circulation research, 2004. 94(12): p. 1543-1553.
[12] Knorr, M., T. Münzel, and P. Wenzel, Interplay of NK cells and monocytes in vascular inflammation and myocardial infarction. Frontiers in physiology, 2014. 5: p. 295.
[13] Ivashkiv, L.B., IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nature Reviews Immunology, 2018. 18(9): p. 545-558.
[14] Shapouri‐Moghaddam, A., et al., Macrophage plasticity, polarization, and function in health and disease. Journal of cellular physiology, 2018. 233(9): p. 6425-6440.

