Atherosclerosis, the pathological underpinning of coronary artery disease (CAD), is a chronic inflammatory disorder in which macrophages exert a central regulatory role.
This study focuses on the function of macrophages in CAD-associated atherosclerotic processes, with specific emphasis on their involvement in plaque inflammation, the mechanisms underlying macrophage-mediated plaque rupture, the correlation between macrophage apoptosis and plaque necrosis, the identification of secreted cytokines that exacerbate plaque instability, and the evaluation of macrophage targeting as a potential strategy to mitigate myocardial infarction risk in CAD patients.
Table of Contents
1. The role of macrophages in inflammation of atherosclerotic plaques
2. How macrophages promote plaque rupture in coronary artery disease?
3. The link between macrophage apoptosis and necrotic core formation in plaques
4. What cytokines do macrophages release that contribute to plaque instability?
5. Macrophage Targeting for the Reduction of Myocardial Infarction Risk in Coronary Artery Disease
01 The role of macrophages in inflammation of atherosclerotic plaques
Macrophages exert a central and multifaceted role in the inflammation of atherosclerotic plaques, acting as key orchestrators from the initial stages of lesion formation to the destabilization and rupture of advanced plaques[1]. Atherosclerosis itself is characterized as a chronic inflammatory state driven by an imbalance in lipid metabolism[2].
The process initiates with the infiltration of circulating monocytes into the subendothelial space of the arterial wall, particularly in regions subjected to disturbed blood flow and endothelial dysfunction. These monocytes differentiate into macrophages, which subsequently internalize oxidized low-density lipoprotein (oxLDL) particles via scavenger receptors such as SR-A and CD36. Unlike the regulated LDL receptor, these scavenger receptors lack an “off switch”, leading to unregulated lipid uptake and the transformation of macrophages into lipid-laden foam cells. These foam cells are a hallmark of early atherosclerotic lesions, known as fatty streaks[3].
Macrophages also exhibit considerable plasticity, responding to microenvironmental cues by polarizing into distinct phenotypes, primarily M1 (pro-inflammatory macrophages) and M2 (anti-inflammatory/reparative) subsets. The balance between these phenotypes is critical for plaque development and stability. M1 macrophages (pro-inflammatory macrophages) are characterized by enhanced lipid uptake, elevated production of pro-inflammatory cytokines, and promotion of plaque progression and instability, which is associated with metabolic reprogramming toward enhanced glycolysis. Key markers for M1 macrophages (pro-inflammatory macrophages) include CD68 and CX3CR1. In contrast, M2 macrophages, generally regarded as atheroprotective, are involved in lipid efflux, inflammation resolution, and tissue repair. However, the role of specific M2 subsets (e.g., CD163+ macrophages) is complex: while historically considered atheroprotective, accumulating evidence supports a pro-atherogenic role in certain contexts[4]. An M1-predominant imbalance contributes to plaque instability. Strategies targeting the M1-to-M2 shift in macrophage polarization are being explored as potential therapeutic interventions for atherosclerosis.
Furthermore, a critical aspect of macrophage-mediated inflammation in atherosclerosis is the activation of the NLRP3 inflammasome. Cholesterol crystals, formed from accumulated lipids in plaques, act as danger signals that trigger NLRP3 inflammasome activation. This activation leads to the cleavage of pro-IL-1β into its mature, highly pro-inflammatory form, IL-1β, which exacerbates the inflammatory response within the plaque and contributes to plaque instability. Additionally, macrophage junctional adhesion molecule-like (JAML) protein has also been shown to promote NLRP3 inflammasome activation in atherosclerosis development[5]. Inhibition of NLRP3 inflammasome, can hinder atherosclerosis progression by attenuating inflammation and pyroptosis in macrophages. Pyroptosis, an intensely inflammatory form of programmed cell death, also contributes to the necrotic core expansion within plaques.
Notably, the metabolic state of macrophages directly modulates their function. M1 macrophages (pro-inflammatory macrophages) typically rely on aerobic glycolysis, whereas M2 macrophages utilize oxidative phosphorylation and fatty acid oxidation. This metabolic reprogramming is intricately intertwined with the atherosclerotic plaque microenvironment. Dysregulated metabolism within macrophages can promote inflammation and foam cell formation. Furthermore, impaired efferocytosis is a critical contributor to chronic inflammation in advanced plaques. When apoptotic foam cells are not efficiently cleared, they undergo secondary necrosis, releasing their cytotoxic contents and expanding the plaque’s necrotic core, which further amplifies inflammation[6].
Collectively, macrophages are pivotal regulators in all stages of atherosclerotic inflammation. Their diverse functions, including lipid uptake, foam cell formation, pro-inflammatory cytokine secretion, activation of inflammasomes, and matrix degradation, collectively drive plaque development, progression, and ultimately, destabilization. Modulating macrophage function and polarization therefore represents a promising therapeutic target for atherosclerosis.
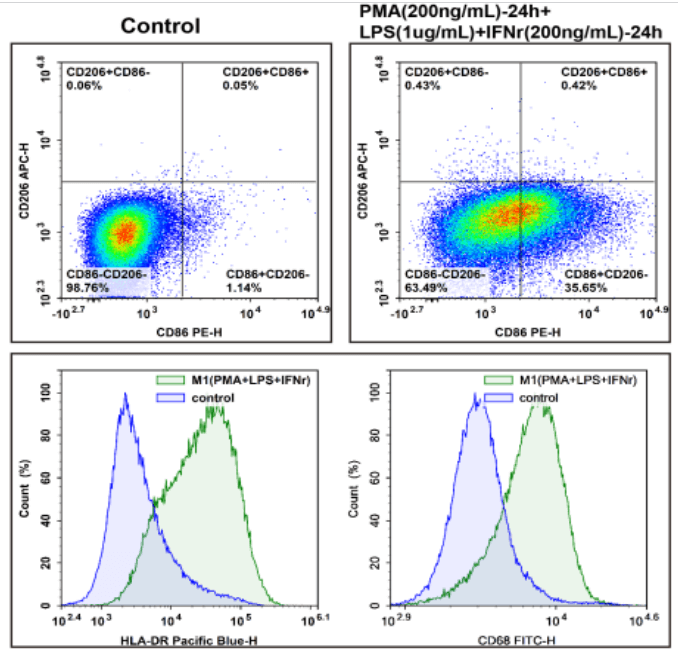
Fig. 1 Analysis of M1 phenotypic polarization in THP-1-derived macrophages. Following the polarization of THP-1 cells toward the M1 phenotype, the expression levels of the indicated cell surface markers were detected and analyzed by flow cytometry using the following fluorescent antibodies: PE‑conjugated CD86 (E-AB-F1012D), APC‑conjugated CD206 (E-AB-F1161E), Elab Fluor® Violet 450‑conjugated HLA-DR (E-AB-F1111Q), Elab Fluor® 488‑conjugated CD68 (E-AB-F1299L), and PerCP/Cyanine5.5‑conjugated CD163 (E-AB-F1298J). Compared with untreated control cells, the M1‑polarized cells displayed significantly elevated expression of HLA-DR, CD86, and CD68, accompanied by markedly reduced expression of CD206. (The datas are provided by Elabceience.)
02 How macrophages promote plaque rupture in coronary artery disease?
Macrophages directly mediate atherosclerotic plaque rupture by actively degrading the structural integrity of plaques and amplifying local inflammatory responses, thereby forming a biomechanically fragile region that is predisposed to tearing under arterial hemodynamic pressure. The key destructive mechanisms by which macrophages drive plaque rupture are summarized as follows:
Destruction of the Fibrous Cap: Macrophages secrete matrix metalloproteinases (MMPs), a family of enzymes that specifically degrade the collagen-rich fibrous cap, leading to progressive thinning of this protective structure[7]. Concomitantly, they suppress the synthesis of new collagen by smooth muscle cells, thereby compromising the reparative capacity of the plaque and exacerbating fibrous cap vulnerability[7].
Expansion of the Necrotic Core: Lipid-laden macrophages undergo necrotic cell death, resulting in the release of their intracellular contents into the plaque microenvironment. Inefficient clearance of these necrotic debris leads to the formation and progressive expansion of a soft, gelatinous necrotic core, which further elevates mechanical stress on the already thinned fibrous cap[8].
Induction of Pathological Angiogenesis: Macrophages promote the ingrowth of fragile neovessels into the plaque parenchyma. These immature neovessels are characterized by poor structural stability and are prone to rupture, triggering intraplaque hemorrhage. This hemorrhage facilitates the recruitment of additional inflammatory cells and lipids into the plaque, which in turn further activates macrophages and amplifies the pathological process[9].
Perpetuation of a Vicious Inflammatory Cycle: Through signaling pathways such as NLRP3 inflammasome activation, macrophages secrete proinflammatory cytokines (e.g., interleukin-1β, IL-1β). These cytokines efficiently recruit and activate additional macrophages into the plaque, establishing a self-perpetuating inflammatory loop that sustains and exacerbates plaque instability[10].
The Final Rupture Trigger: The cumulative effects of a thin, degraded fibrous cap and an enlarged, gelatinous necrotic core render the plaque highly vulnerable to rupture. When arterial hemodynamic stress exceeds the mechanical tolerance of this weakened structure, plaque rupture occurs, exposing thrombogenic components to the systemic circulation and initiating thrombus formation, which ultimately culminates in myocardial infarction[11].
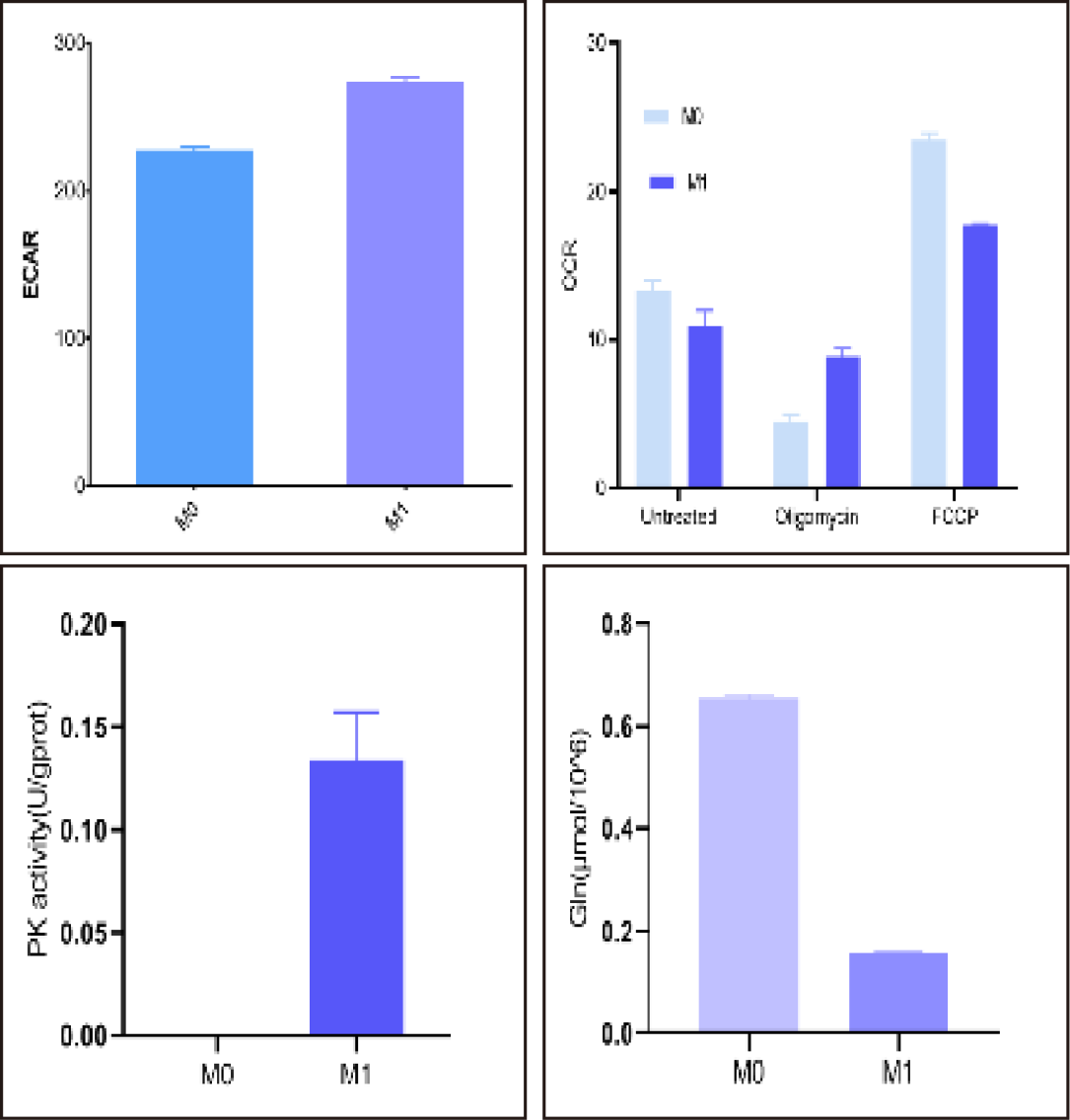
Fig. 2 Detection and analysis of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) in M1-polarized macrophages. Following induction of pyroptosis in RAW264.7 cells via combined treatment with LPS and ATP, cell culture supernatants were collected. LDH release was subsequently detected and quantified using an LDH cytotoxicity assay kit (Cat. No. E-BC-K771-M). Our data demonstrated a significant elevation in LDH release from pyroptotic cells. (The datas are provided by Elabceience.)
03 The link between macrophage apoptosis and necrotic core formation in plaques
Macrophage apoptosis is intricately linked to plaque necrosis in the context of atherosclerosis, a chronic inflammatory vascular disorder, and exerts a pivotal role in regulating plaque vulnerability and progression[12]. Under physiological conditions, billions of cells undergo programmed cell death (apoptosis) daily, and their clearance by phagocytes via a process termed efferocytosis is indispensable for maintaining tissue homeostasis and suppressing inflammation[12]. However, in advanced atherosclerotic plaques, this delicate balance is disrupted, leading to defective efferocytosis and the accumulation of apoptotic cells.
When apoptotic macrophages are not efficiently cleared, they undergo secondary necrosis, a disordered and uncontrolled form of cell death. This secondary necrosis releases pro-inflammatory and pro-thrombotic intracellular contents, including lipids, cholesterol crystals, and danger-associated molecular patterns (DAMPs), into the extracellular space. Such release further fuels inflammation and drives the formation and expansion of the lipid-rich necrotic core, a hallmark of vulnerable atherosclerotic plaques. The presence of a large necrotic core significantly compromises plaque stability, rendering it predisposed to rupture and subsequent acute cardiovascular events (e.g., myocardial infarction, stroke)[13].
Several mechanisms contribute to macrophage apoptosis and the subsequent development of necrotic cores in atherosclerosis. Oxidized low-density lipoprotein (oxLDL) is a major driver of macrophage apoptosis within the plaque[14]. Furthermore, endoplasmic reticulum stress (ERS) and the unfolded protein response (UPR), often triggered by lipid overload, can induce macrophage apoptosis via pathways involving C/EBP-homologous protein (CHOP)[15]. Mitochondrial dysfunction, specifically mediated by cyclophilin D (CypD), has also been identified as a promoter of mitochondria-dependent macrophage apoptosis, thereby facilitating necrotic core formation in advanced lesions. Inflammation-related proteins such as IRGM/Irgm1 can accelerate atherosclerosis progression by inducing macrophage apoptosis through the MAPK signaling pathway. Conversely, interventions that inhibit macrophage apoptosis (e.g., treatment with K-80003) have been shown to attenuate necrotic core expansion and stabilize atherosclerotic plaques. Similarly, Intermedin1-53 has been demonstrated to reduce plaque vulnerability by suppressing CHOP-mediated apoptosis and inflammasome activation in macrophages[15].
Efferocytosis failure, the impaired clearance of apoptotic cells by macrophages, is a critical step linking macrophage apoptosis to necrotic core expansion. This efferocytosis defect can be attributed to multiple factors within the atherosclerotic microenvironment, including impaired “eat-me” signals on apoptotic cells, reduced phagocytic capacity of macrophages, and downregulation of bridging molecules required for engulfment. The persistence of unengulfed apoptotic cells then promotes their progression to secondary necrosis, transforming a non-inflammatory clearance process into a potent inflammatory stimulus[16].
The accumulation of dead cells within the plaque, especially due to inefficient efferocytosis, also creates a unique environment conducive to coronary artery calcification. Apoptotic bodies resulting from these dead cells can serve as nucleation sites for the formation of calcium phosphate crystals, primarily hydroxyapatite, further contributing to plaque progression and instability[17].
Therefore, the relationship between macrophage apoptosis and plaque necrosis is a direct and detrimental one. Macrophage apoptosis, when coupled with defective efferocytosis, directly drives the formation and expansion of the necrotic core, a key feature of vulnerable atherosclerotic plaques that predisposes them to rupture and subsequent acute cardiovascular events. Therapeutic strategies aimed at stabilizing atherosclerotic plaques often focus on either inhibiting macrophage apoptosis or enhancing efferocytosis[18].
Elabscience® Quick Overview of Popular Products:
Table 1. Reagents for Macrophages Research
|
Product Name |
Cat. No. |
|
ATP Colorimetric Assay Kit (Enzyme Method) |
E-BC-K774-M |
|
ATP Chemiluminescence Assay Kit (Double Reagent) |
E-BC-F300 |
|
Caspase 1 Activity Assay Kit(Colorimetric Method) |
E-CK-A381 |
|
Caspase 1 Activity Detection Substrate for Flow Cytometry |
E-CK-A481 |
|
Reactive Oxygen Species (ROS) Fluorometric Assay Kit (Red) |
E-BC-F005 |
|
Lactate Dehydrogenase (LDH) Cytotoxicity Colorimetric Assay Kit |
E-BC-K771-M |
|
Mouse TNF-α(Tumor Necrosis Factor Alpha) ELISA Kit |
E-EL-M3063 |
|
Mouse IL-6(Interleukin 6) ELISA Kit |
E-EL-M0044 |
|
Mouse IL-18(Interleukin 18) ELISA Kit |
E-EL-M0730 |
04 What cytokines do macrophages release that contribute to plaque instability?
Macrophages contribute to plaque instability primarily through the release of pro-inflammatory cytokines that weaken the fibrous cap, promote extracellular matrix degradation, and induce vascular smooth muscle cell (VSMC) apoptosis. The key cytokines involved are shown in the following table.
Table 2. Cytokines and Mediators Released by Macrophages Contributing to Plaque Instability[19,20].
|
Cytokine/Mediator |
Function in Plaque Instability |
|
Tumor necrosis factor-α (TNF-α) |
Promotes inflammation, activates matrix metalloproteinases (MMPs), and weakens the plaque fibrous cap. |
|
Interleukin-1β (IL-1β) |
Amplifies local inflammation, induces endothelial dysfunction, and accelerates plaque progression. |
|
Interleukin-6 (IL-6) |
Enhances systemic inflammation, promotes thrombogenicity, and increases plaque vulnerability. |
|
Interferon-γ (IFN-γ) |
Inhibits collagen synthesis by smooth muscle cells, thinning the protective fibrous cap. |
|
Matrix metalloproteinases (MMPs (e.g., MMP-1, MMP-9)) |
Directly degrade extracellular matrix and further destabilize atherosclerotic plaques. |
05 Macrophage Targeting for the Reduction of Myocardial Infarction Risk in Coronary Artery Disease
Targeted therapy against macrophages in coronary artery disease (CAD) represents a state-of-the-art strategy for reducing the risk of myocardial infarction (MI). The core of this approach lies in modulating the inflammatory behaviors of macrophages within atherosclerotic plaques, promoting plaque stability, and even facilitating plaque regression.
To date, macrophage-targeted therapeutic strategies primarily encompass the following approaches: either inhibiting chemotactic pathways (e.g., CCL2/CCR2) to reduce monocyte recruitment to atherosclerotic plaques at the source, or reprogramming macrophages via pharmacological agents and nanotechnologies to induce a phenotypic switch from the pro-inflammatory M1 subset to the anti-inflammatory, pro-resolving M2 phenotype, thereby alleviating local inflammation and promoting plaque stabilization[21]. Additionally, a pivotal complementary strategy involves enhancing efferocytosis. For instance, targeting the MerTK pathway improves the efficiency of apoptotic cell clearance by macrophages, consequently attenuating the formation of plaque necrotic cores[22].
Notably, clinical breakthroughs have been achieved in directly targeting key inflammatory cascades. IL-1β inhibitors and colchicine, both exerting broad-spectrum anti-inflammatory effects, have been validated in clinical trials to significantly reduce the risk of major adverse cardiovascular events (MACE)[23].
Presently, the most promising frontier of research focuses on advanced drug delivery systems, including ligand-functionalized nanoparticles and biomimetic nanocarriers, which are engineered to precisely deliver therapeutics to plaque-resident macrophages. This targeted delivery maximizes therapeutic efficacy while minimizing systemic adverse effects.
Despite ongoing challenges associated with precise immunomodulation and balancing safety-efficacy profiles, macrophage-targeted therapies have gradually advanced from the conceptual framework of “anti-inflammation” toward partial clinical translation, continually providing innovative strategies for atherosclerotic plaque stabilization[24,25].
References:
[1] Gianopoulos I, Daskalopoulou SS. Macrophage profiling in atherosclerosis: understanding the unstable plaque. Basic Res Cardiol. 2024 Feb;119(1):35-56. doi: 10.1007/s00395-023-01023-z.
[2] Fang F, Xiao C, Li C, Liu X, Li S. Tuning macrophages for atherosclerosis treatment. Regen Biomater. 2022 Dec 13;10:rbac103. doi: 10.1093/rb/rbac103.
[3] Zhang T, Pang C, Xu M, Zhao Q, Hu Z, Jiang X, Guo M. The role of immune system in atherosclerosis: Molecular mechanisms, controversies, and future possibilities. Hum Immunol. 2024 Mar;85(2):110765. doi: 10.1016/j.humimm.2024.110765.
[4] Zhao J, Ling L, Zhu W, Ying T, Yu T, Sun M, Zhu X, Du Y, Zhang L. M1/M2 re-polarization of kaempferol biomimetic NPs in anti-inflammatory therapy of atherosclerosis. J Control Release. 2023 Jan;353:1068-1083. doi: 10.1016/j.jconrel.2022.12.041.
[5] Karasawa T, Takahashi M. The crystal-induced activation of NLRP3 inflammasomes in atherosclerosis. Inflamm Regen. 2017 Sep 11;37:18. doi: 10.1186/s41232-017-0050-9.
[6] Xiao S, Qi M, Zhou Q, Gong H, Wei D, Wang G, Feng Q, Wang Z, Liu Z, Zhou Y, Ma X. Macrophage fatty acid oxidation in atherosclerosis. Biomed Pharmacother. 2024 Jan;170:116092. doi: 10.1016/j.biopha.2023.116092.
[7] Kawai K, Vozenilek AE, Kawakami R, Sato Y, Ghosh SKB, Virmani R, Finn AV. Understanding the role of alternative macrophage phenotypes in human atherosclerosis. Expert Rev Cardiovasc Ther. 2022 Sep;20(9):689-705. doi: 10.1080/14779072.2022.2111301.
[8] De Meyer GRY, Zurek M, Puylaert P, Martinet W. Programmed death of macrophages in atherosclerosis: mechanisms and therapeutic targets. Nat Rev Cardiol. 2024 May;21(5):312-325. doi: 10.1038/s41569-023-00957-0.
[9] Parma L, Peters HAB, Baganha F, Sluimer JC, de Vries MR, Quax PHA. Prolonged Hyperoxygenation Treatment Improves Vein Graft Patency and Decreases Macrophage Content in Atherosclerotic Lesions in ApoE3*Leiden Mice. Cells. 2020 Feb 1;9(2):336. doi: 10.3390/cells9020336.
[10] Kawtharany L, Bessueille L, Issa H, Hamade E, Zibara K, Magne D. Inflammation and Microcalcification: A Never-Ending Vicious Cycle in Atherosclerosis? J Vasc Res. 2022;59(3):137-150. doi: 10.1159/000521161.
[11] De Meyer GRY, Zurek M, Puylaert P, Martinet W. Programmed death of macrophages in atherosclerosis: mechanisms and therapeutic targets. Nat Rev Cardiol. 2024 May;21(5):312-325. doi: 10.1038/s41569-023-00957-0.
[12] De Meyer GRY, Zurek M, Puylaert P, Martinet W. Programmed death of macrophages in atherosclerosis: mechanisms and therapeutic targets. Nat Rev Cardiol. 2024 May;21(5):312-325. doi: 10.1038/s41569-023-00957-0.
[13] Shu LX, Cao LL, Guo X, Wang ZB, Wang SZ. Mechanism of efferocytosis in atherosclerosis. J Mol Med (Berl). 2024 Jul;102(7):831-840. doi: 10.1007/s00109-024-02439-3.
[14] Ning Y, Jia Y, Yang Y, Wen W, Huang M, Liu S, Yang Y, Dong Y, Zhang M. Thyroid hormones inhibit apoptosis of macrophage induced by oxidized low-density lipoprotein. Biofactors. 2022 Jan;48(1):86-99. doi: 10.1002/biof.1803.
[15] Ren JL, Chen Y, Zhang LS, Zhang YR, Liu SM, Yu YR, Jia MZ, Tang CS, Qi YF, Lu WW. Intermedin1-53 attenuates atherosclerotic plaque vulnerability by inhibiting CHOP-mediated apoptosis and inflammasome in macrophages. Cell Death Dis. 2021 May 1;12(5):436. doi: 10.1038/s41419-021-03712-w.
[16] Wang Z, Su J, Gong F, Xue L, Su Z. The Impaired Mechanism and Facilitated Therapies of Efferocytosis in Atherosclerosis. J Cardiovasc Pharmacol. 2022 Sep 1;80(3):407-416. doi: 10.1097/FJC.0000000000001311.
[17] Neels JG, Gollentz C, Chinetti G. Macrophage death in atherosclerosis: potential role in calcification. Front Immunol. 2023 Jul 4;14:1215612. doi: 10.3389/fimmu.2023.1215612.
[18] Tang X, Huang Z, Wang F, Chen J, Qin D, Peng D, Yu B. Macrophage-specific deletion of MIC26 (APOO) mitigates advanced atherosclerosis by increasing efferocytosis. Atherosclerosis. 2023 Dec;386:117374. doi: 10.1016/j.atherosclerosis.2023.117374.
[19] Li S, Zhou X, Duan Q, Niu S, Li P, Feng Y, Zhang Y, Xu X, Gong SP, Cao H. Autophagy and Its Association with Macrophages in Clonal Hematopoiesis Leading to Atherosclerosis. Int J Mol Sci. 2025 Apr 1;26(7):3252. doi: 10.3390/ijms26073252.
[20] Libby P, Geng YJ, Aikawa M, Schoenbeck U, Mach F, Clinton SK, Sukhova GK, Lee RT. Macrophages and atherosclerotic plaque stability. Curr Opin Lipidol. 1996 Oct;7(5):330-5. doi: 10.1097/00041433-199610000-00012.
[21] Wen J, Guan Y, Niu H, Dang Y, Guan J. Targeting cardiac resident CCR2+ macrophage-secreted MCP-1 to attenuate inflammation after myocardial infarction. Acta Biomater. 2024 Aug 23:S1742-7061(24)00469-0. doi: 10.1016/j.actbio.2024.08.025.
[22] Xu Y, Zhang Y, Yu W. Nano-therapeutics targeting the macrophage-based microenvironment in the treatment of atherosclerosis. J Transl Med. 2025 Oct 24;23(1):1171. doi: 10.1186/s12967-025-07222-7.
[23] Fang F, Wang E, Fang M, Yue H, Yang H, Liu X. Macrophage-based pathogenesis and theranostics of vulnerable plaques. Theranostics. 2025 Jan 2;15(4):1570-1588. doi: 10.7150/thno.105256.
[24] Chen Q, Peng YS, Zhong Q, Zhang YB, Hu LB, Zhang GY, Xu YX, Dong PN, Shen S, Wang JH, Wang ZG, Zhang HT, Zeng R, Wang H. Targeted suppression of CCR7/NF-κB signaling by apoptotic body-inspired colchicine nanovesicles halts atherosclerotic progression. J Nanobiotechnology. 2025 Nov 25;23(1):798. doi: 10.1186/s12951-025-03840-x.
[25] Fang F, Wang E, Fang M, Yue H, Yang H, Liu X. Macrophage-based pathogenesis and theranostics of vulnerable plaques. Theranostics. 2025 Jan 2;15(4):1570-1588. doi: 10.7150/thno.105256.

