Ischemic heart disease (IHD), characterized by myocardial ischemia, involves complex interactions between metabolic alterations and immune responses, particularly those orchestrated by T cells[1]. Following ischemic injury, T cells undergo significant metabolic reprogramming, shifting from oxidative phosphorylation (OXPHOS) to aerobic glycolysis, a process driven by key regulators like mTOR and HIF-1α in the hypoxic microenvironment. This glycolytic switch promotes the differentiation of pro-inflammatory subsets such as Th1 and Th17 cells, which exacerbate cardiac injury, while simultaneously impairing the function of reparative regulatory T cells (Tregs) that primarily rely on fatty acid oxidation. The resulting metabolic imbalance sustains inflammation and drives adverse cardiac remodeling.
In this context, we outline the central role of T cell metabolic fate in IHD pathogenesis and its therapeutic potential. Targeting these metabolic pathways, for example by inhibiting glycolysis or enhancing OXPHOS, represents a promising strategy to restore immune homeostasis, suppress maladaptive inflammation, and improve cardiac repair after ischemic injury.
Table of Contents
1. Metabolic reprogramming drives T cell differentiation in ischemic heart disease
2. Key metabolic regulators controlling T cell function during ischemic injury
3. Mitochondrial dysfunction and metabolic shifts in T cells during cardiac ischemia
4. How memory and naive T cells differ metabolically under ischemic stress
5. Experimental models to study T cell metabolic reprogramming in ischemic heart disease
6. Can metabolic interventions restore T cell homeostasis after myocardial infarction?
01 Metabolic reprogramming drives T cell differentiation in ischemic heart disease
Metabolic reprogramming is a key regulator of T cell differentiation in ischemic heart disease (IHD)[2]. Naive T cells primarily depend on OXPHOS for energy homeostasis and undergo marked metabolic reprogramming upon activation and differentiation[3, 4]. Within the ischemic microenvironment (hypoxia and nutrient depletion), T cells preferentially switch to aerobic glycolysis to fulfill the elevated energy requirements for proliferation and effector function[5]. This glycolytic skewing preferentially promotes the differentiation of pro-inflammatory effector T cell subsets (e.g., Th1 and Th17 cells), which contribute to exacerbated cardiac injury and remodeling post-myocardial ischemia[6]. For instance, interferon-gamma (IFN-γ) drives glycolysis-facilitated Th1 cells, thus promoting atherosclerotic plaque destabilization, a key pathological feature of IHD[2].
In contrast, regulatory T cells (Tregs), which are essential for maintaining immune homeostasis and resolving inflammation, primarily depend on fatty acid oxidation(FAO) for their function[1]. A glycolysis-dominant metabolic imbalance impairs Treg differentiation and function, perpetuating inflammation and driving adverse cardiac remodeling[7]. This shift in T cell differentiation toward pro-inflammatory phenotypes can worsen myocardial ischemia symptoms. Metabolic regulators like hypoxia-inducible factor-1α (HIF-1α) stabilize under hypoxia, promoting glycolytic genes and Th17 differentiation, while inhibiting Treg suppressive functions[8].
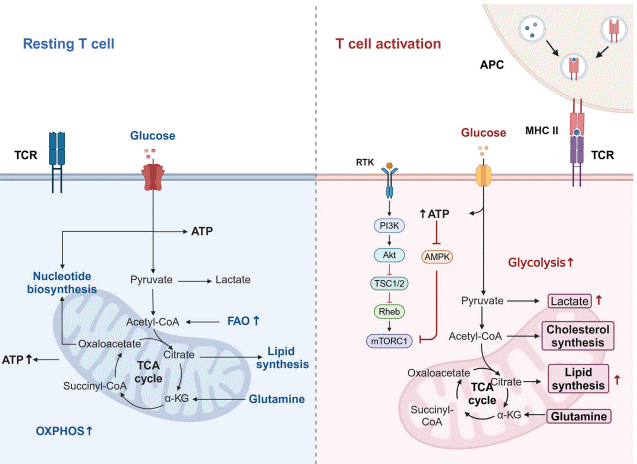
Fig. 1 Metabolic pathway shifts in T cells[4]. T cell activation triggers dynamic metabolic reprogramming, shifting from mitochondrial OXPHOS in resting T cells to enhanced glycolysis in activated cells. This switch supports rapid proliferation and effector functions by meeting heightened energy demands.
02 Key metabolic regulators controlling T cell function during ischemic injury
Several key metabolic regulators orchestrate T cell metabolism in ischemic heart disease.
Mammalian target of rapamycin (mTOR): The mTOR pathway, particularly mTOR complex 1 (mTORC1), is a central regulator that promotes anabolic processes and glycolytic metabolism, driving T cell activation and effector differentiation[9, 10]. Sustained high mTORC1 signaling drives T cells toward a short-lived effector cell fate, whereas reduced mTORC1 activity favors memory T cell differentiation[9, 11]. Akt, activated by PI3K signaling, is a major upstream activator of mTORC1, linking co-stimulation to metabolic reprogramming.
HIF-1α: Hypoxia-inducible factor-1α (HIF-1α) is another critical regulator, particularly active under hypoxic conditions prevalent in ischemic tissues[12]. HIF-1α promotes glycolytic gene expression, further skewing T cells towards a glycolytic phenotype.
AMP-activated protein kinase (AMPK): AMPK, an energy-sensing kinase, is activated when cellular ATP levels are low and promotes catabolic pathways such as FAO and mitochondrial biogenesis, which are characteristic of memory and regulatory T cells[13]. This intricate balance between mTORC1 and AMPK pathways determines the metabolic fate of T cells, influencing their differentiation and function in the context of cardiac ischemia[9].
P2 receptors: Additionally, P2 receptors, modulated by extracellular ATP, can influence T-lymphocyte energy metabolism and function[14]. Mitochondrial metabolism itself serves as a biosynthetic and signaling hub; for example, mitochondrial electron transport chain (ETC) function is essential for T cell activation through ROS signaling, and metabolites like succinate or L-2-hydroxyglutarate can modulate epigenetic programs in T cells[6].
03 Mitochondrial dysfunction and metabolic shifts in T cells during cardiac ischemia
Mitochondrial dysfunction and metabolic reprogramming are tightly interconnected in T cells during cardiac ischemia. Myocardial ischemia is characterized by reduced blood flow and oxygen supply, leading to impaired mitochondrial function in cardiomyocytes and infiltrating immune cells[15]. Mitochondria are responsible for approximately 90% of cellular ATP production through OXPHOS. During ischemia, dysfunctional mitochondria exhibit reduced membrane potential, increased ROS production, and impaired electron transport chain activity, contributing to cellular injury[16]. In T cells, this mitochondrial dysfunction can impair their ability to generate ATP efficiently via OXPHOS, forcing them to rely more heavily on glycolysis, even in the presence of oxygen[5]. Although this metabolic shift enables rapid ATP production, it is less energy-efficient and induces lactate accumulation, which further aggravates the acidic and immunosuppressive microenvironment. Impaired mitochondrial activity also affects naive T cell differentiation and memory T cell differentiation by limiting the spare respiratory capacity (SRC) essential for long-term survival and function. The interaction between mitochondrial dysfunction and metabolic shifts forms a feedback loop, sustaining inflammation and hindering tissue repair in ischemic heart disease. For instance, in atherosclerotic plaques, hypoxia and oxidized lipids induce mitochondrial ROS in macrophages, which further promotes pro-inflammatory T cell responses[15].
04 How memory and naive T cells differ metabolically under ischemic stress
Memory T cells exhibit metabolic adaptations that confer enhanced resilience to ischemic stress relative to naive T cells[17]. The table below summarizes their key differential characteristics.
Table 1. Metabolic Characteristics of Naive vs. Memory T Cells in Response to Ischemic Stress
| Feature |
Naive T Cells |
Memory T Cells |
|
Primary Metabolic Pathway |
OXPHOS |
Fatty Acid Oxidation (FAO) & OXPHOS; greater metabolic flexibility[18] |
|
Mitochondrial Fitness |
low spare respiratory capacity (SRC) |
Higher SRC[18] |
|
Intracellular Fuel Reserves |
Limited stores |
Maintain glycogen and triacylglyceride stores for energy |
|
Response to Ischemia |
Energetic collapse due to high sensitivity to hypoxia and nutrient deprivation |
Sustained ATP production via metabolic switching (e.g., to FAO), enhanced survival |
|
Key Molecular Regulators |
Sensitivity to mTOR inhibition |
PPARβ/δ[2], enhanced mitochondrial biogenesis |
The enhanced survival of memory T cells under ischemic stress is attributed to their mitochondrial fitness and metabolic flexibility. They maintain a high spare respiratory capacity, allowing them to rapidly generate energy in response to stress. This metabolic advantage allows memory T cells to survive and function effectively in the challenging microenvironment of an ischemic tissue, such as the heart after a myocardial infarction. While this is crucial for mounting a rapid immune response, it can also sometimes contribute to maladaptive inflammation, influencing the progression of conditions like ischemic heart disease.
05 Experimental models to study T cell metabolic reprogramming in ischemic heart disease
The investigation of T-cell metabolic reprogramming in ischemic heart disease relies on diverse models, including in vivo murine models, ex vivo systems, and human-based platforms. These approaches elucidate critical metabolic pathways like glycolysis, oxidative phosphorylation, and fatty acid oxidation, which fundamentally govern T-cell fate and cardiac results.
5.1 In Vivo Murine Models
Myocardial Infarction (MI) Models: Permanent coronary artery ligation in mice induces ischemic injury, recruiting T cells to the heart. This model reveals time-dependent metabolic adaptations: pro-inflammatory Th1 and Th17 cells favor glycolysis, while reparative Tregs rely on mitochondrial OXPHOS[19, 20].
Ischemia-Reperfusion (I/R) Injury: Transient coronary occlusion mimics clinical revascularization. I/R promotes HIF-1α-driven glycolytic shifts in T cells and amplifies mitochondrial stress, which can be modulated by interventions like CD73 inhibition or metabolic preconditioning[19].
Pressure Overload Models: Transverse aortic constriction (TAC) induces hypertrophy and chronic inflammation, highlighting the role of metabolic regulators (e.g., PHD2 and HIF-1α) in T-cell glycolysis and dysfunction[19].
5.2 Ex Vivo and Human-Based Platforms
Adoptive T-Cell Transfer: Transfer of metabolically labeled T cells into IHD models tracks their fate in vivo. For example, fatty acid-preconditioned CD4+ T cells exhibit upregulated lipid metabolism, altering differentiation toward pro-inflammatory subsets[19, 21].
HiPSC-Derived Cardiomyocyte Models: Human-induced pluripotent stem cell (hiPSC)-derived cardiomyocytes exposed to hypoxia/glucose deprivation simulate ischemic conditions[22, 23]. Co-culture with T cells allows study of human-specific immunometabolic crosstalk.
Humanized Mice: Immunodeficient mice engrafted with human T cells enable exploration of human metabolic pathways in cardiac injury[19].
5.3 Approaches for Metabolic Analysis
Metabolomics/Lipidomics: Mass spectrometry identifies metabolic intermediates in T cells isolated from IHD models.
Microplate reader: Measures glycolytic flux (ECAR) and OXPHOS (OCR) in infiltrating cardiac T cells.
Flow Cytometry: Metabolic tracer techniques profile single-cell metabolism in T-cell subsets.
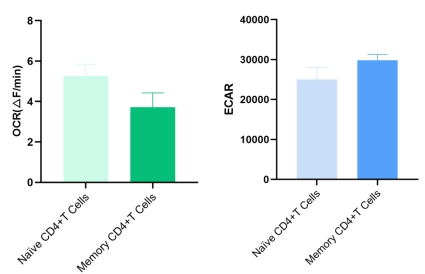
Fig. 2 Measurement of OCR and ECAR in human naive and memory CD4+ T cells. Naive and memory CD4+ T cells were sorted from human PBMCs. OCR and ECAR were measured using specific assay kits. Memory CD4+ T cells exhibited increased ECAR compared to naive CD4+ T cells (The datas are provided by Elabscience).
06 Can metabolic interventions restore T cell homeostasis after myocardial infarction?
In the progression of ischemic heart disease, sustained reduction in coronary blood flow often culminates in acute myocardial infarction (MI), Following MI, maladaptive T cell metabolism contributes to sustained inflammation and impaired cardiac repair. Notably, myocardial ischemia treatment (including revascularization, pharmacotherapy, and metabolic regulation) aims to alleviate tissue hypoxia and restore cardiac function, among which metabolic reprogramming of T cells emerges as a novel therapeutic target. Metabolic reprogramming represents a promising therapeutic strategy to rebalance T cell responses and promote homeostasis. Key mechanisms and interventions include:
Targeting Glycolytic Pathways:
Glycolysis inhibition (e.g., via 2-deoxyglucose) attenuates pro-inflammatory Th17 and effector T cell activity, while promoting anti-inflammatory Treg differentiation[19]. Sodium-glucose cotransporter 2 (SGLT2) inhibitors (e.g., empagliflozin) reprogram CD4+ T cell metabolism by switching energy dependence from glycolysis to FAO, thereby enhancing Treg-mediated inflammatory suppression[24].
Enhancing Oxidative Metabolism:
Activation of AMPK promotes OXPHOS and fatty acid oxidation, supporting memory T cell and Treg development[19, 25]. This metabolic shift dampens excessive inflammatory responses and improves cardiac outcomes.
Purinergic Signaling and Immunomodulation:
CD73 on T cells converts pro-inflammatory ATP to adenosine, thereby activating A2a/A2b receptors to suppress the production of IFN-γ and IL-17. This inhibition alleviates excessive post-infarction inflammation, leading to reduced myocardial tissue damage[26] CTLA-4-Ig therapy (e.g., abatacept) inhibits T cell co-stimulation, reducing infarct expansion and preserving function[27].
Quick Overview of Popular Products:
Table 2. Reagents for T Cells Metabolic Reprogramming and Ischemic Heart Disease Research
|
Cat. No. |
Product Name |
|
E-CK-A103 |
Human PBMC Separation Solution (P 1.077) |
|
E-CK-A091 |
Cell Stimulation and Protein Transport Inhibitor Kit |
|
E-CK-A109 |
Intracellular Fixation/Permeabilization Buffer Kit |
|
E-CK-A105 |
10×ACK Lysis Buffer |
|
MIM002N |
EasySort™ Mouse CD4+T Cell Isolation Kit |
|
MIM003N |
EasySort™ Mouse CD8+T Cell Isolation Kit |
|
MIM001N |
EasySort™ Mouse CD3+T Cell Isolation Kit |
|
MIH001N |
EasySort™ Human CD3+T Cell Isolation Kit |
|
MIH002N |
EasySort™ Human CD4+ T Cell Isolation Kit |
|
MIH003N |
EasySort™ Human CD8+ T Cell Isolation Kit |
|
MIH001A |
Human CD3/CD28 T Cell Activation Beads |
|
MIM001A |
Mouse CD3/CD28 T Cell Activation Beads |
|
E-CK-A108 |
Foxp3/Transcription Factor Staining Kit |
|
E-BC-F070 |
Enhanced Oxygen Consumption Rate (OCR)Fluorometric Assay Kit |
|
E-BC-F069 |
Extracellular Acidification Rate (ECAR) Fluorometric Assay Kit |
|
E-BC-F201 |
Enhanced ATP Chemiluminescence Assay Kit |
|
E-BC-F084 |
Glycolysis Stress Fluorometric Assay Kit |
|
E-BC-K784-M |
Fatty Acid Oxidation (FAO) Colorimetric Assay Kit |
References:
[1] Li, M., et al., Metabolic Homeostasis of Immune Cells Modulates Cardiovascular Diseases. Research, 2025. 8.
[2] Chang, S., Z. Wang, and T. An, T-Cell Metabolic Reprogramming in Atherosclerosis. Biomedicines, 2024. 12(8): p. 1844.
[3] Gerriets, V.A. and J.C. Rathmell, Metabolic pathways in T cell fate and function. Trends in Immunology, 2012. 33(4): p. 168-173.
[4] Shi, Y., H. Zhang, and C. Miao, Metabolic reprogram and T cell differentiation in inflammation: current evidence and future perspectives. Cell Death Discov, 2025. 11(1): p. 123.
[5] Zhang, S., et al., Hurdle or thruster: Glucose metabolism of T cells in anti-tumour immunity. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer, 2024. 1879(1): p. 189022.
[6] Steinert, E.M., K. Vasan, and N.S. Chandel, Mitochondrial Metabolism Regulation of T Cell-Mediated Immunity. Annu Rev Immunol, 2021. 39: p. 395-416.
[7] Lu, Y., et al., Immunometabolism of Tregs: mechanisms, adaptability, and therapeutic implications in diseases. Front Immunol, 2025. 16: p. 1536020.
[8] Dang, E.V., et al., Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell, 2011. 146(5): p. 772-84.
[9] Huang, J., S. Leary, and J. Xiang, Distinct strengths of mTORC1 control T-cell memory via transcriptional FOXO1 and metabolic AMPKα1 pathways in linear cell differentiation and asymmetric cell division models. Cellular & Molecular Immunology, 2022. 19(10): p. 1073-1076.
[10] Delgoffe, G.M., et al., The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nature Immunology, 2011. 12(4): p. 295-303.
[11] Araki, K., et al., mTOR regulates memory CD8 T-cell differentiation. Nature, 2009. 460(7251): p. 108-112.
[12] Li, H., et al., Metabolic Adaptations and Therapies in Cardiac Hypoxia. JACC: Basic to Translational Science, 2025. 10(6): p. 862-878.
[13] Escrig-Larena, J.I., S. Delgado-Pulido, and M. Mittelbrunn, Mitochondria during T cell aging. Seminars in Immunology, 2023. 69: p. 101808.
[14] Vultaggio-Poma, V. and F. Di Virgilio, P2 Receptors: Novel Disease Markers and Metabolic Checkpoints in Immune Cells. Biomolecules, 2022. 12(7): p. 983.
[15] Schirone, L., et al., An Overview of the Molecular Mechanisms Associated with Myocardial Ischemic Injury: State of the Art and Translational Perspectives. Cells, 2022. 11(7): p. 1165.
[16] Crola Da Silva, C., et al., Isolated Mitochondria State after Myocardial Ischemia-Reperfusion Injury and Cardioprotection: Analysis by Flow Cytometry. Life, 2023. 13(3): p. 707.
[17] Metur, S.P. and D.J. Klionsky, Adaptive immunity at the crossroads of autophagy and metabolism. Cellular & Molecular Immunology, 2021. 18(5): p. 1096-1105.
[18] Mallat, Z. and C.J. Binder, The why and how of adaptive immune responses in ischemic cardiovascular disease. Nature Cardiovascular Research, 2022. 1(5): p. 431-444.
[19] Mouton, A.J. and J.E. Hall, Novel roles of immunometabolism and nonmyocyte metabolism in cardiac remodeling and injury. Am J Physiol Regul Integr Comp Physiol, 2020. 319(4): p. R476-r484.
[20] Bansal, S.S., et al., Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circulation: Heart Failure, 2017. 10(3): p. e003688.
[21] Chang, S., Z. Wang, and T. An, T-Cell Metabolic Reprogramming in Atherosclerosis. Biomedicines, 2024. 12(8).
[22] Ye, J., et al., Effect and mechanism of T lymphocytes on human induced pluripotent stem cell-derived cardiomyocytes via Proteomics. Stem Cell Research & Therapy, 2024. 15(1): p. 236.
[23] Wei, H., et al., Development of a model of ischemic heart disease using cardiomyocytes differentiated from human induced pluripotent stem cells. Biochemical and Biophysical Research Communications, 2019. 520(3): p. 600-605.
[24] Canonico, F., et al., GLUT-1/PKM2 loop dysregulation in patients with non-ST-segment elevation myocardial infarction promotes metainflammation. Cardiovasc Res, 2023. 119(16): p. 2653-2662.
[25] Braverman, E.L., et al., Overexpression of AMPKγ2 increases AMPK signaling to augment human T cell metabolism and function. J Biol Chem, 2024. 300(1): p. 105488.
[26] Borg, N., et al., CD73 on T Cells Orchestrates Cardiac Wound Healing After Myocardial Infarction by Purinergic Metabolic Reprogramming. Circulation, 2017. 136(3): p. 297-313.
[27] Noonan, J., et al., CTLA-4-Ig therapy preserves cardiac function following myocardial infarction with reperfusion. Cardiovasc Res, 2025. 121(13): p. 2082-2094.

