Interferons (IFNs) are key regulators of innate and adaptive immunity and are classified into three types based on receptor usage and structural features: type I (including IFN-α, IFN-β, etc.), type II (IFN-γ), and type III (IFN-λ). This review focuses on IFN-α, IFN-β, and IFN-γ, systematically comparing interferons function and signaling mechanisms. IFN-α primarily induces broad-spectrum antiviral proteins via the ifn JAK-STAT pathway and exerts immunomodulatory effects during viral infections. IFN-β possesses dual antiviral and immunoregulatory functions in innate immunity and inflammation and serves as a first-line therapy for multiple sclerosis. IFN-γ plays a central role in adaptive immunity by activating macrophages and driving Th1 differentiation. The three IFN types differ fundamentally in receptor composition, JAK kinase preference, STAT complex formation, and downstream transcriptional response elements (ISRE vs. GAS), which determine their distinct gene expression profiles and biological functions. Furthermore, advances in IFN quantification using ELISA and multiplex assays (e.g., Simoa, Luminex, FluoroSpot) provide essential technical support for precise assessment of IFN activity and clinical therapeutic guidance.
Table of Contents
1. Overview of interferon families and classification
2. Interferon-alpha (IFN-α): antiviral signaling and immune modulation
3. Interferon-beta (IFN-β): role in innate immunity and inflammation
4. Interferon-gamma (IFN-γ): regulation of adaptive immunity
5. Differences in signaling pathways among IFN-α, IFN-β, and IFN-γ
6. Measurement of IFN levels using ELISA and multiplex assays
01 Overview of interferon families and classification
IFNs, first discovered by Isaacs and Lindenmann in 1957, have since evolved into a complex cytokine family comprising multiple subtypes as core mediators of innate immunity. Based on differences in molecular structure, receptor specificity, and signaling mechanisms, IFNs are systematically classified into three major types: type I, type II, and type III, each playing a distinct role in host defense and disease therapy[1].
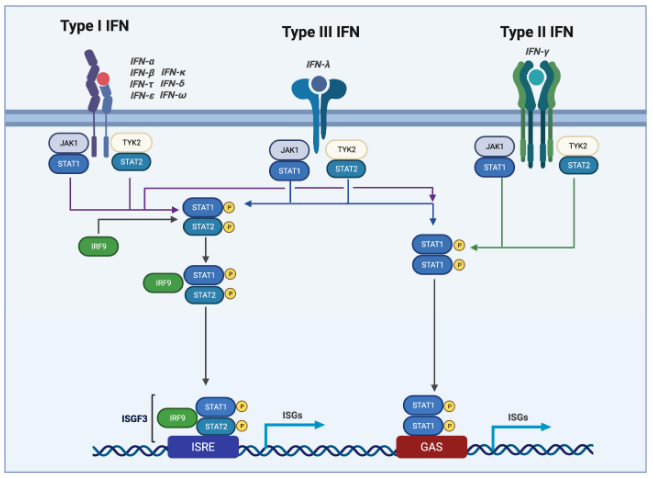
Fig. 1 Type 1, type II, and type III IFN signaling[1]. Type I, II, and III IFNs all trigger JAK1 and TYK2 activation, resulting in STAT phosphorylation. Phosphorylated STAT1 forms heterodimers with STAT2 and recruits IRF9 to generate the ISGF3 complex, which translocates to the nucleus, binds ISRE sequences, and induces ISG expression.
Type I IFNs constitute the largest subtype family, primarily including IFN-α (13 subtypes), IFN-β, IFN-ε, IFN-κ, and IFN-ω. They bind to the type I interferon receptor (IFNAR), composed of IFNAR1 and IFNAR2, on the cell surface to activate the JAK-STAT signaling pathway, thereby inducing the expression of hundreds of antiviral proteins, such as Mx proteins, 2'-5' oligoadenylate synthetase (OAS), and protein kinase R (PKR), thus establishing a broad-spectrum antiviral state[2]. Type I interferons are primarily produced by plasmacytoid dendritic cells (pDCs), fibroblasts, and epithelial cells, enabling rapid responses to viral infections and activating immune defense mechanisms in neighboring cells in a paracrine manner. Recent studies have revealed functional differences among type I interferon subtypes; for instance, IFN-α14 exhibits more potent antiviral activity than IFN-α2 in the treatment of chronic hepatitis B (CHB)[3].
Type II IFN comprises only one member, IFN-γ, which is primarily produced by activated T lymphocytes, natural killer (NK) cells, and innate lymphoid cells (ILCs). IFN-γ activates the ifn JAK-STAT pathway by binding to the type II interferon receptor (IFNGR), composed of IFNGR1 and IFNGR2, thereby inducing the expression of immunomodulatory genes, enhancing MHC molecule expression and antigen presentation capacity, and serving as a key bridge connecting innate and adaptive immunity[4].
Type III IFNs (also known as IFN-λ or the IL-28/29 family) were first identified in 2003 and comprise four subtypes: IFN-λ1 (IL-29), IFN-λ2 (IL-28A), IFN-λ3 (IL-28B), and IFN-λ4. Type III IFNs exert their effects by binding to the type III interferon receptor (IFNLR), composed of IFNLR1 and IL-10RB, and are mainly expressed in epithelial cells and mucosal tissues, exhibiting a characteristic of mucosal-specific defense. Compared with type I interferons, type III IFNs provide antiviral protection while inducing milder systemic inflammatory responses, making them a new hotspot for investigating targeted immune interventions. Studies have demonstrated that IFN-λ drives diarrhea through activation of the ifn JAK-STAT pathway during enteric viral infections, thereby facilitating pathogen clearance, revealing a novel mechanism in mucosal immunity[5].
02 Interferon-alpha (IFN-α): antiviral signaling and immune modulation
IFN-α, a core member of the type I interferon family, plays a dual role in antiviral defense and immune regulation. Since its first discovery in 1957, IFN-α has become a key signaling molecule bridging innate and adaptive immunity and is widely used in the treatment of viral infections (e.g., hepatitis C, COVID-19) and cancer immunotherapy.
The antiviral signaling of IFN-α is primarily mediated through two core pathways: the JAK-STAT pathway and the interferon regulatory factor (IRF) pathway. A study demonstrated that upon binding to the IFNAR1/2 on the cell surface, IFN-α activates JAK1 and TYK2 kinases, which subsequently phosphorylate STAT1 and STAT2 to form the STAT1/2-IRF9 trimeric complex (ISGF3). This complex translocates to the nucleus and binds to interferon-stimulated response elements (ISRE), inducing the expression of hundreds of interferon-stimulated genes (ISGs). Concurrently, viral nucleic acids activate the IRF3/7 pathway via pattern recognition receptors (PRRs) such as TLR7/9 or RIG-I/MDA5. Studies have shown that upon recognition of viral single-stranded RNA by TLR7, the MyD88-dependent pathway activates the TBK1/IKKε kinase complex, which then phosphorylates IRF7 to induce the expression of IFN-α subtypes[6].
IFN-α not only suppresses viral replication by directly inducing antiviral proteins but also exerts immunomodulatory effects through regulating various immune cells. In antiviral immunity, IFN-α promotes DC maturation via the ifn JAK-STAT pathway, enhances their antigen-presenting capacity, and upregulates the expression of co-stimulatory molecules (e.g., CD80/CD86), thereby facilitating T cell activation. Studies have shown that IFNα2b can restore the function of dendritic cells from patients with the JAK2v617f mutation by inhibiting STAT3 phosphorylation and downregulating FGL2 expression, thereby enhancing antitumor immune responses[7]. The regulation of B-cell function by IFN-α also represents a critical component of its immunomodulatory role. Studies have demonstrated that IFN-α coordinates protective HBV-specific cellular immunity in a B-cell-dependent manner. The underlying mechanism involves enhanced B-cell–T-cell interactions mediated by antigen presentation and co-stimulatory signals, particularly through promoting B-cell activation and antibody class switching to strengthen humoral immune responses[8]. Furthermore, IFN-α exerts a biphasic regulatory effect on NK cell functions. IFN-α can activate the cytotoxic function of immature NK cells, enhance their killing activity against target cells, and promote the upregulation of surface molecules such as CD69, CD11b, and CD2, while also stimulating IFN-γ secretion. However, it inhibits IL-2-mediated NK cell proliferation and TNF-α secretion[9]. This regulatory mechanism suggests that IFN-α plays a finely balanced role between immune cell activation and suppression.
03 Interferon-beta (IFN-β): role in innate immunity and inflammation
IFN-β, a key member of the type I interferon family, is primarily produced by fibroblasts and plasmacytoid dendritic cells (pDCs) upon pathogen infection. It triggers the JAK-STAT signaling cascade by binding to the type I interferon receptor IFNAR on the cell surface, inducing the expression of hundreds of interferon-stimulated genes (ISGs), thereby establishing an antiviral state and regulating inflammatory responses.
The induction and signal transduction of IFN-β constitute the first line of defense in innate immunity. During viral infection, pathogen-associated molecular patterns (PAMPs) are recognized by host pattern recognition receptors (PRRs), which subsequently activate transcription factors IRF3, IRF7, and NF-κB to drive IFN-β transcription[10]. Furthermore, the RIG-I-MAVS pathway plays a critical role in RNA virus recognition. A recent study revealed that p53 upregulates the hexosamine biosynthesis pathway to promote O-GlcNAcylation of UBXN1 and MAVS, thereby enhancing IFN-β-mediated antiviral innate immunity[11]. Precise regulation of IFN-β expression is essential, as overproduction can lead to immunopathological damage. One study demonstrated that the E3 ubiquitin ligase RAD18 specifically recognizes phosphorylated and activated IRF3, promoting its dissociation from the IFNB1 promoter via K63-linked ubiquitination and subsequent degradation through the autophagy-lysosome pathway, thereby effectively curbing excessive IFN-β expression[12]. Additionally, the SP140-RESIST pathway regulates IFN-β expression at the level of mRNA stability: RESIST prolongs the half-life of Ifnb1 mRNA by counteracting TTP-mediated mRNA destabilization, whereas SP140 limits IFN-β production by inhibiting RESIST transcription[13].
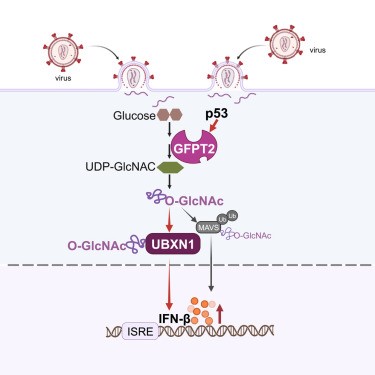
Fig. 2 p53 enhances IFN-β-mediated antiviral innate immunity by regulating the hexosamine biosynthesis pathway and O-GlcNAc modification[11]. p53 metabolically enhances antiviral innate immunity by transcriptionally upregulating GFPT2 to drive the hexosamine biosynthetic pathway and O-GlcNAcylation of UBXN1 and MAVS, which releases MAVS to activate TBK1-IRF3 signaling and IFN-β production. Genetic or pharmaceutical inhibition of GFPT abrogates this p53-promoted antiviral response.
The role of IFN-β in antiviral innate immunity has also been extensively studied. pDCs, as professional type I interferon-producing cells, exhibit heterogeneous subpopulations even under steady-state conditions. Recent studies have shown that within resting pDCs, the “IFN-I-naive subpopulation” (pDC-A) displays a higher capacity for IFN-I production but is more susceptible to viral infection. In contrast, pDC-B/C subpopulations that have experienced tonic IFN-I signaling exhibit reduced IFN-I responses but acquire enhanced resistance to viral infection. This finding reveals the central regulatory role of IFN-I signaling in the functional differentiation of pDCs[14]. The role of IFN-β in bacterial infection is more complex, exhibiting both protective and detrimental effects. Type I interferons can exert protective functions during infections with Mycobacterium tuberculosis, Listeria monocytogenes, and others; however, in the context of other pathogens, excessive IFN-β responses paradoxically exacerbate host damage. This double-edged sword effect is highly dependent on specific factors such as pathogen species, route of infection, and host genetic background[15]. In a sepsis model, systemic inflammation induces a state of long-term immunosuppression by impairing monocyte maturation and type I interferon signaling, whereas IFN-β treatment can restore myeloid function and reverse immunosuppression, offering a novel strategy for immune reconstitution after sepsis[16].
The regulation of inflammatory responses by IFN-β reflects its dual immunomodulatory nature. On one hand, IFN-β induces the expression of pro-inflammatory ISGs during antiviral processes; on the other hand, it exerts anti-inflammatory effects through multiple mechanisms. In a 2025 review, Brunsting and colleagues proposed that IFN-β mediates immune regulation via transcriptional repression, including suppression of the IL-17 and Th17 pathways[17]. Regarding NLRP3 inflammasome regulation, studies have shown that memory T cells pre-treated with IFN-β inhibit NLRP3 inflammasome activation by downregulating P2X7R signaling. Moreover, this inhibitory capacity is impaired in patients with multiple sclerosis (MS) but is restored following IFN-β therapy[18].
In the field of MS treatment, significant breakthroughs have been made in recent years regarding the mechanism of action of IFN-β. Studies have revealed that IFN-β inhibits the secretion of miR-21-inducing cytokines by myeloid cells, thereby blocking miR-21-mediated differentiation of pathogenic Th17 cells. In IFN-β non-responders, the expression of miR-21 and its inducing cytokines is significantly elevated, suggesting that miR-21 inhibition may represent a potential therapeutic target for this patient population[19]. Concurrently, using multiplex proteomic analysis, Li et al. found that IFN-β treatment corrects dysregulated levels of interferon-stimulated proteins, serum cytokines, and neurotrophic factors in MS patients, restoring the immune environment to a balanced state[20]. Furthermore, IFN-β therapy has shown neuroprotective potential in ischemic stroke models by promoting the polarization of microglia toward an anti-inflammatory phenotype and maintaining blood–brain barrier integrity. However, the clinical application of IFN-β faces the challenge of neutralizing antibody development. Persistently present neutralizing antibodies completely block the biological activity of IFN-β, and continued administration offers no therapeutic benefit once neutralizing antibody titers become elevated[21].
04 Interferon-gamma (IFN-γ): regulation of adaptive immunity
IFN-γ, discovered in 1965, is the sole member of the type II interferon family and is primarily produced by activated T cells, NK cells, and ILCs. Th1 differentiation represents a central process through which IFN-γ regulates adaptive immunity. Leonard et al. identified Aiolos as a positive regulator of IFN-γ/STAT1 signaling; Aiolos-deficient CD4+ T cells exhibit reduced expression of JAK2 and STAT1, leading to impaired STAT1 phosphorylation and diminished CXCR3 transcription, and the positive feedback loop formed between Aiolos and STAT1 further consolidates Th1 lineage commitment[22]. Recent studies have revealed that glycolysis stabilizes intracellular IFN-γ production through OGT‑mediated O‑GlcNAcylation of STAT1, thereby driving Th1 effector function[23]. Moreover, the distal silencer CNS‑28 in the Ifng locus suppresses IFN‑γ transcription in a GATA3‑dependent manner, and its deletion exacerbates autoimmunity[24]. In the tumor microenvironment, IFN‑γ drives the differentiation of Tregs into Th1‑like effector Tregs, which inhibit DC1‑mediated CTL priming via MHC class II‑dependent contact, thereby impairing antitumor immunity[25]. Excessive IFN‑γ released by CAR‑T cells can induce cytokine release syndrome, whereas blockade of IFN‑γ reduces toxicity while preserving therapeutic efficacy[26].
ifn gamma macrophage activation via the JAK/STAT1-IRF1 axis, promoting phagolysosome maturation, production of reactive oxygen and nitrogen species, LC3-associated phagocytosis, and autophagy. Mycobacterium tuberculosis and Mycobacterium leprae evade this defense axis through mechanisms such as receptor downregulation, induction of IL-10/SOCS, and type I interferon antagonism[27]. Imran et al. emphasize that host-directed therapy requires balancing the protective and detrimental effects of IFN-γ. Gorin et al. discovered that the persistence of IFN-γ signaling alone is sufficient to maintain the formation of de novo enhancers in macrophages, conferring long-lasting transcriptional memory to subsequent stimuli independent of other extrinsic factors[28]. Murphy et al. demonstrated that IFN-γ induces trained immunity in monocytes through mTORC1 activation, glutaminolysis, and epigenetic remodeling; ATAC‑seq revealed significantly increased chromatin accessibility at host defense‑related regions in IFN‑γ‑trained macrophages, and IFN‑γ training restored impaired innate immune responses in individuals carrying the TIRAP 180L susceptibility polymorphism[29].
05 Differences in signaling pathways among IFN-α, IFN-β, and IFN-γ
5.1 Receptor composition and binding kinetics
Type I IFNs (including IFN-α and IFN-β) share the heterodimeric receptor IFNAR1/IFNAR2, which is expressed on the surface of nearly all nucleated cells. The different biological effects mediated by IFN-α and IFN-β through the same receptor arise from the combined effects of ligand-receptor binding kinetics and cell surface receptor density. In receptor-scarce cells, the antiproliferative activity of IFN-β is significantly stronger than that of IFN-α2, whereas in receptor-abundant cells, IFN-α2 exhibits comparable or even superior activity. This finding challenges the traditional notion that the same receptor mediates identical signals. Type II IFN (IFN-γ), in contrast, utilizes a distinct receptor, IFNGR1/IFNGR2, whose expression profile is more selective for immune cells[30].
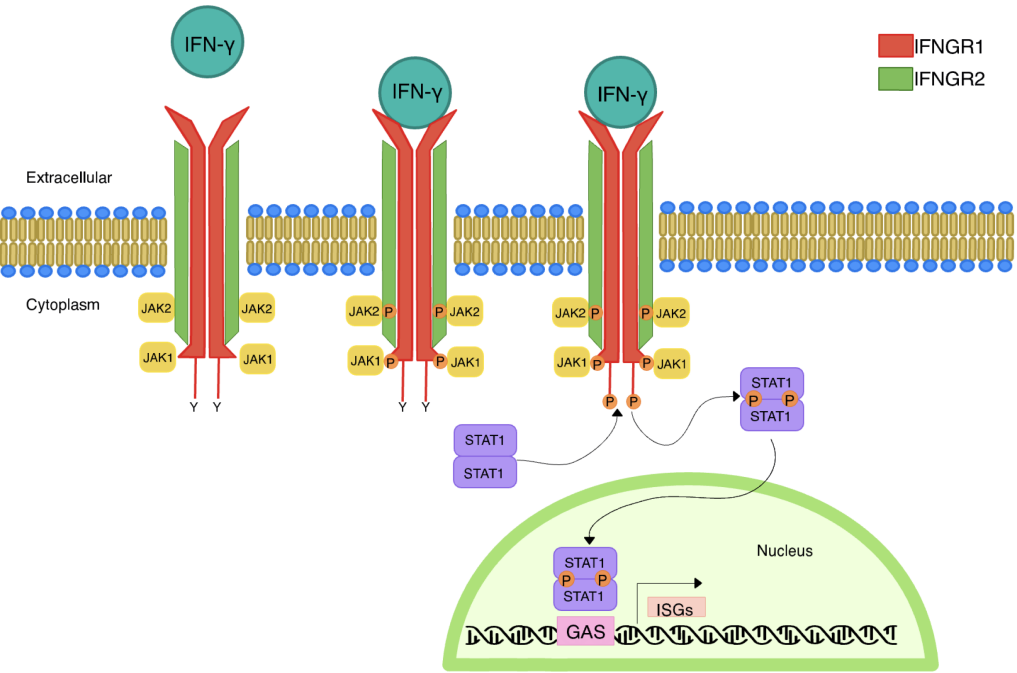
Fig. 3 Canonical signaling pathway of IFN-γ[30]. The heterotetrameric receptor complex of IFN-γ is composed of IFNGR1 and IFNGR2. JAK1 and JAK2 associate with the intracellular domains of IFNGR1 and IFNGR2, respectively. Binding of IFN-γ to its receptor complex promotes trans-phosphorylation of JAK1/2, phosphorylation of IFNGR1, and recruitment of STAT1, followed by phosphorylation of STAT1 by JAK proteins. Phosphorylated STAT1 translocates into the nucleus and regulates the transcription of genes containing gamma-activated sequence motifs in their promoter regions.
5.2 JAK kinases and STAT complexes
Type I IFNs activate JAK1 and TYK2, leading to phosphorylation of STAT1 and STAT2, which assemble with IRF9 into the ISGF3 trimer[31]. Type II IFN activates JAK1 and JAK2, forming the STAT1 homodimer (GAF)[30]. A study by Zoler et al. revealed that JAK1, JAK2, and TYK2 competitively bind to the IFNAR1 and IFNAR2 subunits, and the efficacy of different JAKs in inducing cross-phosphorylation and downstream signaling varies, a competitive binding mechanism that may explain cell type‑dependent signaling pleiotropy[2].
5.3 Downstream transcriptional response elements and gene expression profiles
ISGF3 binds to ISRE elements, driving the expression of broad-spectrum antiviral genes such as MX1, OAS, IFIT, and ISG15. GAF binds to GAS elements, driving the expression of immunomodulatory genes such as NOS2, CIITA, GBP, and CXCL9/10/11, which are particularly associated MHC antigen presentation and inflammatory chemotaxis. Although the two pathways share certain ISGs, the gene programs induced by each exhibit clear biases.
In summary, type I IFNs (particularly IFN-β) are characterized by broad-spectrum antiviral and antiproliferative functions and are widely used in the treatment of multiple sclerosis. Type II IFN (IFN-γ), on the other hand, centers on immunomodulation and macrophage activation, playing a critical role in antitumor immunity, albeit with a “double-edged sword” effect: enhancing antigen presentation and T cell killing while also promoting immune evasion through the induction of molecules such as PD-L1 and IDO1. Moreover, complex cross-regulation exists between type I and type II IFNs: type I IFNs can suppress IFN-γ‑mediated activation of macrophages, whereas type II IFN can enhance the expression of type I IFN‑induced antiviral genes.
06 Measurement of IFN levels using ELISA and multiplex assays
Since the 1970s, ELISA has been the gold standard for cytokine quantification, such as for interferon assay, yet it has long faced the critical bottleneck of insufficient sensitivity. The physiological concentrations of different IFN subtypes in circulation vary considerably: IFN-γ can be reliably quantified in healthy human serum by high‑sensitivity ELISA, whereas type I and type III IFNs, present at extremely low levels, typically fall below the detection limit under standard assay conditions. So the ifn alpha elisa and ifn beta elisa are limited. To address this challenge, research over the past five years has driven technological advances from multiple directions.
First, conventional ifn gamma elisa remains important for IFN‑γ detection. In 2025, a competitive ELISA (cELISA) based on monoclonal antibodies was reported for detecting neutralizing anti‑IFN‑γ autoantibodies (nAIGAs), providing a practical alternative to cell‑based assays for the diagnosis of adult‑onset immunodeficiency (AOID)[32]. However, a comparative evaluation of commercial ifn alpha elisa quantification revealed that low sensitivity and poor reliability mainly stem from the low affinity and specificity of the monoclonal antibody pairs, compounded by the extremely low concentration of IFN‑α in serum, which makes direct quantification even more difficult[33].
The introduction of ultrasensitive technologies has greatly transformed the detection capability for type I IFNs. Simoa (Single‑Molecule Array) digital ELISA technology exhibits the highest sensitivity and precision, with a sensitivity approximately 5,000‑fold greater than that of conventional ELISA. A cross‑sectional observational study used Simoa to accurately quantify IFN‑α and IFN‑γ in serum from 313 SLE patients and found that IFN‑α was significantly positively correlated with inflammatory markers, disease activity index, and autoantibody status, whereas no such association was observed for IFN‑γ[34]. Another study further indicated that Simoa digital ELISA correlates well with the transcriptome‑based IFN‑stimulated gene (ISG) score, and that in SLE the ISG score is primarily explained by circulating IFN‑α levels, with negligible contributions from IFN‑β or IFN‑γ[33].
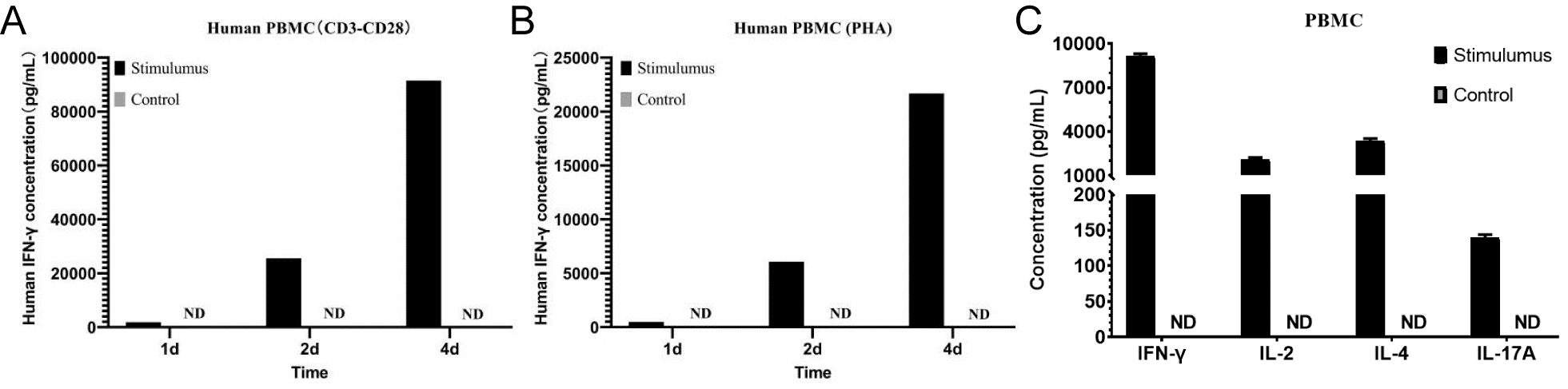
Fig. 4 Detection of IFN-γ levels in the supernatant of human PBMCs by ELISA. A.Human PBMCs were stimulated with 10 μg/mL anti-CD3 mAb + 1 μg/mL anti-CD28 mAb for 1, 2, or 4 days, and IFN-γ levels in the supernatant were measured compared with unstimulated cells; B. Human PBMCs were stimulated with 10 μg/mL PHA for 1, 2, or 4 days, and IFN-γ levels in the supernatant were measured compared with unstimulated cells; C. Human PBMCs (1×10⁶ cells/mL) were left untreated or stimulated with 10 μg/mL PHA + 10 ng/mL PMA, or with PMA (50 ng/mL) + ionomycin (500 ng/mL) for 1 day. The levels of IFN-γ, IL-2, IL-4, and IL-17A in the cell culture supernatant were determined by ELISA. (The data are provided by Elabscience.)
The novel ultrasensitive immunoassay based on S-Plex (electrochemiluminescence) has further enhanced detection performance. One study compared the ability of three technologies to detect serum IFN-β in SLE patients and found that the detection rates of circulating IFN-β by Simoa, ELISA, and S-Plex were 7.5%, 18.8%, and 98.3%, respectively, and in healthy controls were 1.3%, 6.7%, and 100%, respectively. Although the concentration of IFN-β detected by S-Plex was significantly higher in active SLE patients than in inactive patients (0.389 pg/mL vs. 0.243 pg/mL) and correlated with disease severity and remission status, its diagnostic performance in distinguishing clinical disease states was suboptimal, with considerable overlap in levels between patient groups[35].
In addition to ultrasensitive immunoassays, a dual-mode lateral flow aptamer assay based on surface-enhanced Raman scattering (SERS) has been reported to enable accurate quantification of IFN-γ within a dynamic range of 5–2000 pg/mL, achieving a detection limit of 2.23 pg/mL. Combined with machine learning (multinomial logistic regression), this method achieved an overall accuracy of 94.12%, providing a new avenue for the clinical translation of ultrasensitive cytokine detection[36].
Multiplex detection technologies enable the simultaneous analysis of multiple IFN types and other cytokines. The FluoroSpot technique, based on the ifn gamma elispot principle, allows the concurrent detection of the secretion of several cytokines at the single-cell level. One study evaluated a multiplex FluoroSpot assay for the simultaneous detection of IFN-γ, IL-2, and TNF secretion. By identifying triple cytokine-secreting T cells, this approach effectively reduced the false-positive rate and revealed that different stages of tuberculosis infection exhibit distinct cytokine secretion profiles: patients with active tuberculosis had a higher proportion of IFN-γ/TNF-secreting cells, whereas latently infected individuals predominantly harbored IFN-γ/IL-2 or IL-2/TNF-secreting cells[37].
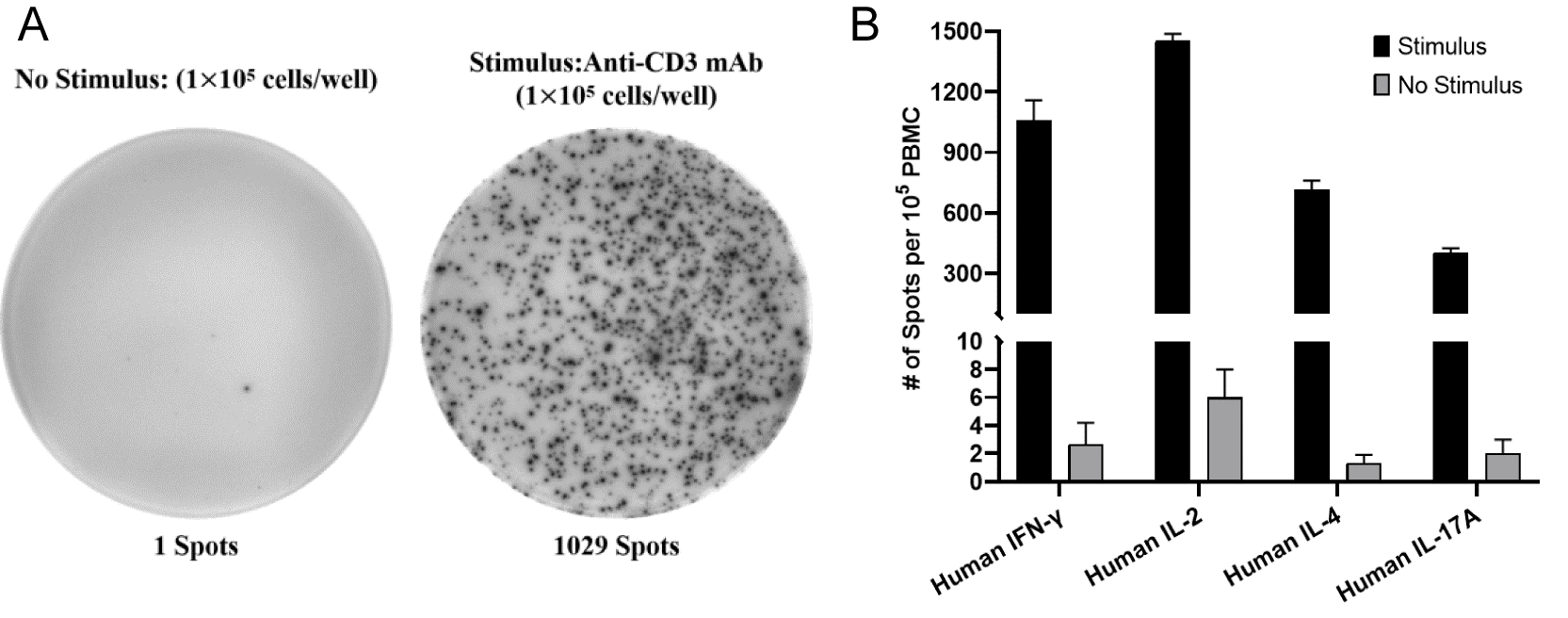
Fig. 5 Detection of IFN-γ expression by ELISpot. A. Human PBMCs (1×10⁵ cells/well) were incubated for 20 h without or with the stimulator anti-CD3 mAb (1:500), The spots represent the number of IFN-γ-secreting cells; B. ELISpot assessment of T cell function. PBMCs were incubated for 20 h without or with the stimulator anti-CD3 mAb (500 ng/mL), and the expression of T cell‑related factors IFN-γ, IL-2, IL-4, and IL-17A was detected by ELISpot. (The data are provided by Elabscience.)
Bead‑based multiplex immunoassays (e.g., the Luminex platform) enable the simultaneous quantification of up to dozens of cytokines in a single sample. A study used the Luminex xMAP platform to perform multiplex quantitative analysis of 15 human cytokines, chemokines, and growth factors in primary human cells[38]. The flow cytometry‑based LegendPlex platform similarly allows simultaneous quantification of 13 human or mouse cytokines, including IFN‑γ, multiple interleukins, and TNF‑α, providing an efficient tool for comprehensive analysis of immune responses mediated by distinct T‑cell subsets such as Th1, Th2, Th9, and Th17[39].
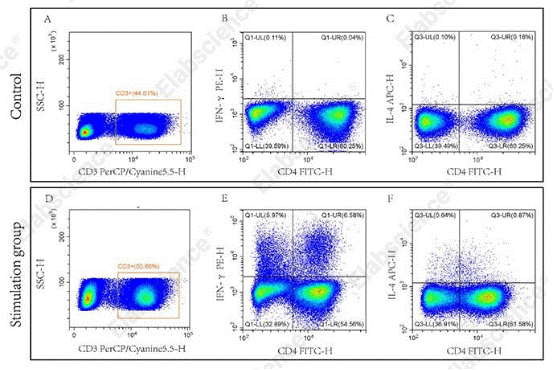
Fig. 6 Flow cytometric analysis was performed using the Mouse Th1/Th2 Flow Cytometry Staining Kit. Splenocytes from normal C57 mice (Control) and from C57 mice stimulated with Cell Stimulation MIX (Stimulation group) were analyzed. CD3⁺CD4⁺IFN-γ⁺ cells (Q1-UR) were identified as Th1 cells, and CD3⁺CD4⁺IL-4⁺ cells (Q3-UR) as Th2 cells. (The data are provided by Elabscience.)
However, current research faces three major challenges. First, although ultrasensitive detection technologies (Simoa, S-Plex) can detect trace amounts of type I IFNs, the dynamic association between their measured values and clinical disease activity requires further validation through longitudinal studies. Second, standardization and quality control systems for multiplex assay platforms remain non‑uniform, and the comparability of results across different platforms needs to be strengthened. Third, the inconsistency between mRNA‑based ISG scores and direct protein quantification results suggests the need for integrated algorithms to achieve more accurate assessment of interferon activity. Future efforts should focus on developing portable multiplex IFN detection devices suitable for point‑of‑care testing (POCT), establishing subtype‑specific quantitative standards for IFNs to eliminate inter‑kit variability, and extending ultrasensitive detection technologies to the clinical diagnosis of other low‑abundance cytokines and interferon‑signaling‑related diseases.
Quick Overview of Popular Products:
Table 1. Reagents and Kits for IFNs
|
Cat. No. |
Product Name |
|
CQH003 |
CellaQuant™ Human IFN-γ (Interferon Gamma) ELISA Kit |
|
CQM005 |
CellaQuant™ Mouse IFN-γ (Interferon Gamma) ELISA Kit |
|
CQH017 |
CellaQuant™ Human IFN-β (Interferon Beta) ELISA Kit |
|
E-EL-M0048 |
Mouse IFN-γ (Interferon Gamma) ELISA Kit |
|
E-EL-H0108 |
Human IFN-γ (Interferon Gamma) ELISA Kit |
|
E-EL-M0033 |
Mouse IFN-β (Interferon Beta) ELISA Kit |
|
E-EL-H0085 |
Human IFN-β (Interferon Beta) ELISA Kit |
|
E-EL-H6125 |
Human IFNα (Interferon Alpha ) ELISA Kit |
|
E-EL-M3054 |
Mouse IFNα (Interferon Alpha) ELISA Kit |
|
E-HSEL-H0007 |
High Sensitivity Human IFN-γ (Interferon Gamma) ELISA Kit |
|
E-HSEL-M0007 |
High Sensitivity Mouse IFN-γ (Interferon Gamma) ELISA Kit |
|
E-MSEL-M0007 |
Mini Sample Mouse IFN-γ (Interferon Gamma) ELISA Kit |
|
E-EL-R0009 |
Rat IFN-γ (Interferon Gamma) ELISA Kit |
|
ESP-M0001 |
Mouse IFN-γ (Interferon Gamma) ELISPOT Kit |
|
ESP-H0002 |
Human IFN-γ (Interferon Gamma) ELISPOT Kit |
|
E-CK-A441 |
Fatty Acid Uptake Fluorometric Assay Kit |
|
E-AB-F1101D |
PE Anti-Mouse IFN-γ Antibody[XMG1.2] |
|
E-AB-F1101A |
Purified Anti-Mouse IFN-γ Antibody[XMG1.2] |
|
AN007590P |
Purified Anti-Human IFN-γ Antibody[4S.B3] |
|
E-AB-F11960 |
AF/LE Purified Anti-Human IFN-γ Antibody[B27] |
|
PDMH100462 |
Recombinant Human IFN-β Protein(Halo Tag) |
|
PDEM100198 |
Recombinant Mouse IFN-γ Protein(GST Tag) |
|
XJH001 |
Human Th1/Th2 Flow Cytometry Staining Kit |
References:
[1] STINE L, CAHILL S, HUMPHRIES F. Interferons in human inborn errors of disease [J]. mBio, 2025, 16(8): e01570-25.
[2] ZOLER E, MEYER T, BELLóN J S, et al. Promiscuous Janus kinase binding to cytokine receptors modulates signaling efficiencies and contributes to cytokine pleiotropy [J]. Science Signaling, 2024, 17(863): eadl1892.
[3] ASHUO A, LIU J, YUAN Z, CHEN J. Interferon-α for immune modulation in chronic hepatitis B toward functional cure [J]. Viruses, 2025, 17(10):1358.
[4] LIU X. The paradoxical role of IFN-γ in cancer: Balancing immune activation and immune evasion [J]. Pathology - Research and Practice, 2025, 272: 156046.
[5] HOU G, SON J, GOMEZ CASTRO M F, et al. Innate immune sensing of rotavirus by intestinal epithelial cells leads to diarrhea [J]. Cell Host & Microbe, 2025, 33(3): 408-19.e8.
[6] ZHU L, JI J, XIAO J, et al. Interferons in HIV-1 infection: mechanisms, antiviral potentials, and therapeutic challenges [J]. Frontiers in Immunology, 2025, 16(1664-3224): 1736658.
[7] FANG L, FU R, DONG H, et al. IFNα2b modulates anti-tumor immune responses involving STAT3-associated dendritic cell dysfunction in JAK2v617f-positive myeloproliferative neoplasms [J]. Drug Resistance Updates, 2026, 85: 101352.
[8] ZHONG S, LI Q, WEN C, et al. Interferon α facilitates anti-HBV cellular immune response in a B cell-dependent manner [J]. Antiviral Research, 2022, 207: 105420.
[9] JEWETT A, BONAVIDA B. Interferon-α activates cytotoxic function but inhibits interleukin-2-mediated proliferation and tumor necrosis factor-α secretion by immature human natural killer cells [J]. Journal of Clinical Immunology, 1995, 15(1): 35-44.
[10] MUHAMMAD I, CONTES K, BILITY M T, TANG Q. Chasing Virus Replication and Infection: PAMP-PRR Interaction Drives Type I Interferon Production, Which in Turn Activates ISG Expression and ISGylation [J].Viruses, 2025, 17(4):528.
[11] XIA W, JIANG P. p53 promotes antiviral innate immunity by driving hexosamine metabolism [J]. Cell Reports, 2024, 43(2): 113724.
[12] CAI Y, ZHENG J, ZHAO L, et al. E3 ligase RAD18 targets phosphorylated IRF3 to terminate IFNB1 transcription [J]. Nature Immunology, 2025, 26(9): 1581-95.
[13] WITT K C, DZIULKO A, AN J, et al. SP140–RESIST pathway regulates interferon mRNA stability and antiviral immunity [J]. Nature, 2025, 643(8074): 1372-80.
[14] PUCELLA J N, MAQUEDA-ALFARO R A, NI H, et al. Tonic type I interferon signaling optimizes the antiviral function of plasmacytoid dendritic cells [J]. Nature Immunology, 2025, 26(11): 1946-61.
[15] XIA A, LI X, ZHAO C, et al. For Better or Worse: Type I Interferon Responses in Bacterial Infection [J]. Pathogens, 2025, 14(3):229.
[16] KERAMATI F, STUNNENBERG HG. Interferon-β treatment restores myeloid function and reverses immunosuppression in sepsis [J]. Nature Immunology, 2025, 26(5): 663-4.
[17] BRUNSTING E A-O X, PERKINS D A-O. Working in negative space: Type I interferon mediated immuno-modulation through transcriptional suppression in disease and homeostasis [J]. Innate Immunity, 2025, 31(1753-4267): 1574-83.
[18] BEYNON V, QUINTANA F J, WEINER H L. Activated human CD4+CD45RO+ memory T-cells indirectly inhibit NLRP3 inflammasome activation through downregulation of P2X7R signalling [J]. PLOS ONE, 2012, 7(6): e39576.
[19] VARGHESE J F, FUJIWARA M, AJAY A K, et al. Type I interferon limits central nervous system autoimmunity by modulating the microRNA-21–FOXO1 axis in pathogenic T helper 17 cells [J]. Science Translational Medicine, 2025, 17(815): eadp5802.
[20] LI L, OLCER M, WANG Z, et al. IFN-β therapy rescues dysregulated IFN-stimulated proteins, serum cytokines, and neurotrophic factors in multiple sclerosis: Multiplex analysis of short-term and long-term IFN responses [J]. PLOS ONE, 2025, 20(9): e0330867.
[21] FITZGERALD S. Antibodies block effect of interferon on MS [J]. Neurology Today, 2009, 9(15): 1, 14.
[22] LEONARD M R, JONES D M, READ K A, et al. Aiolos promotes CXCR3 expression on Th1 cells via positive regulation of IFN-γ/STAT1 signaling [J]. JCI Insight, 2024, 10(1): e180287.
[23] ABIR A A-O, BENZ J, FREY B, et al. Glycolytic flux sustains human Th1 identity and effector function via STAT1 glycosylation. [J]. Life Science Alliance, 2025, 9(1): e2025033.
[24] CUI K, CHEN Z, CAO Y, et al. Restraint of IFN-γ expression through a distal silencer CNS-28 for tissue homeostasis [J]. The Journal of Immunology, 2023, 56(5): 944-58.
[25] CARNEY M, ZAGORULYA M, SPRANGER S. 818 Th1-like tregs suppress the CD8+ T cell response to non-small cell lung cancer [J]. Journal for ImmunoTherapy of Cancer, 2025, 13(Suppl 2).
[26] ZHANG P, YING P, LI H, et al. A novel safer CD19CAR with shRNA interference of IFN-γ can reduce multiple cytokine levels without significantly compromising its killing efficacy [J]. Apoptosis, 2024, 29(3): 556-67.
[27] IMRAN M, ALSHRARI A S, KHAN A, ALZAHRANI A R. IFN-γ-Driven macrophage responses in the immunity to Mycobacterium tuberculosis and Mycobacterium leprae [J]. Frontiers in Cellular and Infection Microbiology, 2025, 15(15): 1679691.
[28] GORIN A, NIU S, HARRIOTT N, et al. IFNγ-induced memory in human macrophages is sustained by the durability of cytokine signaling itself [J]. Journal of Experimental Medicine, 2026, 223(4): e20250976.
[29] MURPHY D M, BATTEN I, O'FARRELL A, et al. IFN-γ-induced trained immunity enhances killing of priority pathogens in healthy and genetically vulnerable individuals. [J]. JCI Insight, 2026, 11(6): e195866.
[30] TECALCO-CRUZ A C, MEDINA-ABREU K H, OROPEZA-MARTíNEZ E, et al. Deregulation of interferon-gamma receptor 1 expression and its implications for lung adenocarcinoma progression [J]. World Journal of Clinical Oncology, 2024, 15(2): 195-207.
[31] MUHAMMAD I, CONTES K, BILITY M T, TANG Q A-O. Chasing virus replication and infection: PAMP-PRR interaction drives type I interferon production, which in turn activates ISG expression and ISGylation. [J]. Viruses, 2025, 17(4): 528.
[32] SORNSUWAN K, THONGHEANG K, WONGSAWAT E, et al. Development of a monoclonal antibody-based competitive ELISA as a surrogate assay for detecting neutralizing anti-interferon gamma autoantibodies in adult-onset immunodeficiency [J]. PLOS ONE, 2026, 21(3): e0344451.
[33] HALJASMäGI L, MEISALU S, BONDET V A-O, et al. Ultrasensitive test systems are required to quantify interferon alpha protein in serum samples [J]. European Journal of Immunology, 2025, 55(12): e70100.
[34] GONZáLEZ-GAY MÁ A-O, GóMEZ-BERNAL F, QUEVEDO-ABELEDO J C, et al. Ultrasensitive quantification of serum IFN-α and IFN-γ in systemic lupus erythematosus: A cross-sectional observational study [J]. PLoS Medicine, 2025, 22(12): e1004841.
[35] DORGHAM K, DA MATA JARDIN O, MELLOT C, et al. Serum interferon-beta level determined by an ultrasensitive electrochemiluminescence immunoassay is increased in clinically active and inactive systemic lupus erythematosus [J]. European Cytokine Network, 2025, 36(2): 15-23.
[36] JIN J, HU J, YAN J A-O, et al. Dual-Mode SERS Lateral Flow Aptamer Assay with Machine Learning-Driven Highly Sensitive Interferon-γ Detection [J]. ACS Synthetic Biology, 2025, 14(7): 2845-53.
[37] FOLKESSON E A-O, FOROOGH F, KLEBERG L A-O, et al. A multiplex Mtb-specific FluoroSpot assay measuring IFNγ, IL-2, and TNF-secreting cells can improve accuracy and differentiation across the tuberculosis spectrum [J]. Journal of Clinical Microbiology, 2025, 63(12): e0089425.
[38] BANETE A, GRIFFIN B D, CORREDOR J C, et al. Pathogenesis and transmission of SARS-CoV-2 D614G, Alpha, Gamma, Delta, and Omicron variants in golden hamsters [J]. npj Viruses, 2025, 3(1): 15.
[39] BIOLEGEND. New LEGENDplex™ Multi-Analyte Flow Assay Panels for Simultaneous Quantification of 13 Th Cytokines in Human and Mouse Samples [Z]. 2014.

