OT-1 cells are a well-established CD8+ T cell line isolated from OT-1 transgenic mice. Their antigen recognition is restricted to MHC class I molecules, with specific reactivity against ovalbumin (OVA). These cells carry a transgenic T cell receptor (TCR), rendering the vast majority of the cell population CD8+ cytotoxic T lymphocytes (CTLs) that stably recognize the ovalbumin peptide spanning residues 257-264. Given these characteristics, OT-1 cells have been widely applied in immunological investigations to explore CD8+ T cell functions, as well as for the development and validation of tumor immunotherapies.
This review systematically summarizes metabolic reprogramming associated with the progression of OT-1 T cell exhaustion. We also discuss the detection of impaired cytotoxicity in exhausted OT-1 CD8+ T cells, compare exhaustion-related molecular signatures between OT-1 and polyclonal CD8+ T cells, and profile the expression dynamics of programmed death-1 (PD-1), T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3) and lymphocyte activation gene 3 (LAG-3) throughout OT-1 T cell expansion. In addition, we outline cytokine supplementation strategies for maintaining the functional competence of OT-1 CD8+ T cells, as well as the selection principles of antigen-presenting cells (APCs) in OT-1 co-culture systems (T cell culture). The commonly used APCs include splenocytes, bone marrow-derived dendritic cells (BMDCs) and artificial APCs (artificial antigen presenting cells).
Table of Contents
1. Metabolic reprogramming during OT-1 T cell exhaustion development
2. Measuring cytotoxic function loss in exhausted OT-1 CD8+ T cells
3. Comparing exhaustion signatures between OT-1 and polyclonal CD8+ T cells
4. Monitoring PD-1, TIM-3, and LAG-3 expression during OT-1 T cell expansion
5. Cytokine supplementation strategies for functional maintenance of OT-1 CD8+ T cells and current research gaps
6. APC selection in OT-1 co-culture models: splenocytes, BMDCs, or artificial APCs
01 Metabolic reprogramming during OT-1 T cell exhaustion development
Metabolic reprogramming during OT-1 CD8+ T cell exhaustion represents a fundamental shift in cellular bioenergetics that determines the functional fate of CD8+ T cells under chronic antigen stimulation. The OT-1 transgenic mouse model, which harbors a T cell receptor (TCR) specific for the ovalbumin-derived peptide SIINFEKL, serves as a powerful tool to delineate the temporal dynamics and molecular mechanisms driving this metabolic dysfunction. T cell exhaustion is not merely a state of functional anergy, but rather an active differentiation program accompanied by stage-specific metabolic alterations. During this process, T cells transit from an effector phenotype dominated by glycolysis to a state marked by severe mitochondrial impairment and metabolic deficiency[1]. This metabolic shift is driven by persistent TCR activation, engagement of inhibitory receptors, and immunosuppressive cues within the tumor microenvironment (TME), which ultimately establish an epigenetically locked exhausted phenotype[2].
Early activation of OT-1 T cells initiates a metabolic switch toward aerobic glycolysis, which is sustained by mammalian target of rapamycin complex 1 (mTORC1) signaling to meet the biosynthetic demands of robust cell proliferation. However, prolonged antigen exposure abolishes such metabolic flexibility[3]. Persistent stimulation induces sustained upregulation of inhibitory receptors including programmed death-1 (PD-1), which potently suppresses the phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K-Akt-mTOR) axis and consequently reduces glucose uptake and glycolytic activity[3]. Unlike acutely activated effector T cells that maintain high glycolytic flux, exhausted OT-1 T cells undergo progressive declines in both glycolysis and oxidative phosphorylation (OXPHOS), leading to a cellular energy crisis that impairs cytokine secretion and cytotoxic granule release. Metabolic insufficiency is a core driver of the hierarchical loss of effector functions in exhausted T cells: interleukin-2 (IL-2) production is lost first, followed by tumor necrosis factor-α (TNF-α) and ultimately interferon-γ (IFN-γ)[4].
Mitochondrial dysfunction acts as a central regulator of OT-1 T cell exhaustion progression. In the hypoxic niche characteristic of solid tumors, chronic TCR triggering elicits severe mitochondrial stress, manifested as fragmented mitochondrial networks, reduced mitochondrial membrane potential and impaired electron transport chain activity. Such mitochondrial damage is not a passive outcome of cellular senescence, but an active process driven by persistent signaling cascades and metabolic stress. The resultant accumulation of reactive oxygen species (ROS) further aggravates cellular injury and stabilizes the exhausted state[4]. Accumulating evidence demonstrates that mitochondrial dysfunction facilitates the transition from precursor exhausted T cells (Tpex) to terminally exhausted T cells (Tex) via hypoxia-inducible factor-1α (HIF-1α)-dependent glycolytic reprogramming. Tpex cells retain intact mitochondrial function and primarily rely on fatty acid oxidation (FAO) and OXPHOS, thereby sustaining self-renewal capacity and responsiveness to immune checkpoint blockade. In contrast, Tex cells develop profound mitochondrial defects and become dependent on aberrant glycolysis, which permanently locks them into a terminally dysfunctional state[5].
The TME further reshapes metabolic profiles of exhausted OT-1 T cells via nutrient competition and accumulation of immunosuppressive metabolites. Tumor cells and stromal components outcompete T cells for essential nutrients including glucose and amino acids, generating a nutrient-deprived microenvironment that compromises T cell metabolic fitness. In addition, lactate, a major byproduct of tumor glycolysis, accumulates extensively in the TME and directly impairs T cell function. Lactate uptake mediated by monocarboxylate transporter 11 (MCT11) sustains T cell dysfunction, linking extracellular metabolic stress to intracellular signaling networks that maintain the exhausted phenotype. Hypoxia, another key feature of the TME, stabilizes HIF-1α to suppress mitochondrial respiration and enforce a glycolytic phenotype incapable of supporting long-term T cell survival and function[6].
Epigenetic modifications are tightly coupled with metabolic alterations and contribute to the stable maintenance of T cell exhaustion. Metabolic intermediates function as essential cofactors for epigenetic enzymes, indicating that the perturbed metabolism of exhausted T cells directly remodels chromatin landscapes. For example, acetyl-coenzyme A (acetyl-CoA) and S-adenosylmethionine (SAM), two key metabolites derived from core metabolic pathways, modulate histone acetylation and methylation, respectively. These epigenetic changes establish a persistent transcriptional program governed by master regulators such as thymocyte selection-associated high mobility group box protein (TOX) and nuclear receptor subfamily 4 group A member (NR4A), which upregulate inhibitory receptors and repress effector-associated genes. Accordingly, the epigenetic "scars" of exhaustion often persist even after metabolic improvement, rendering the exhausted phenotype partially irreversible unless combined metabolic and epigenetic interventions are applied[7].
Current therapeutic strategies to reverse OT-1 T cell exhaustion predominantly focus on metabolic restoration. Enhancing mitochondrial integrity via peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) overexpression or antioxidant treatment can partially recover T cell effector function. Moreover, immune checkpoint blockade represented by anti-PD-1 antibodies not only relieves inhibitory signaling but also restores metabolic capacity by rescuing glycolysis and mitochondrial activity. Targeting distinct metabolic pathways, such as MCT11 inhibition to block lactate uptake or adenosine monophosphate-activated protein kinase (AMPK) activation to boost FAO, provides additional strategies to improve T cell persistence and antitumor efficacy. Elucidating the key metabolic checkpoints governing the Tpex-to-Tex transition is critical for developing next-generation immunotherapies capable of preserving T cell function within the hostile microenvironments of chronic infection and malignancy[8,9].
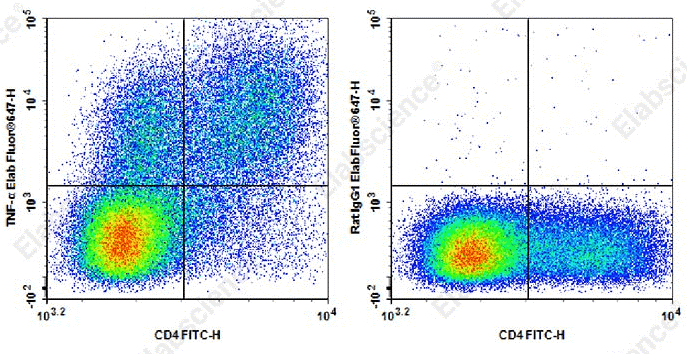
Fig. 1 Detection and analysis of TNF-α in C57BL/6 mouse splenocytes. C57BL/6 mouse splenocytes were stimulated with Cell Stimulation MIX and Protein Transport Inhibitor MIX for 5 h. Cells were stained with FITC anti-mouse CD4 antibody, alongside Elab Fluor® 647 Rat IgG1,κ isotype control (left) or Elab Fluor® 647 anti-mouse TNF-α (XT3.11) (right). Analysis was restricted to lymphocyte-gated cells. (The data are provided by Elabscience.)
Elabscience® Quick Overview of Popular Products:
Table 1. Reagents for research on OT-1 CD8+ T cell exhaustion and metabolism
|
Product Name |
Cat. No. |
|
Elab Fluor® 647 Anti-Mouse TNFα Antibody[XT3.11] |
AN00567M |
|
FITC Anti-Mouse CD4 Antibody[RM4-5] |
E-AB-F1353C |
|
Cell Stimulation and Protein Transport Inhibitor Kit |
E-CK-A091 |
|
Reactive Oxygen Species (ROS) Fluorometric Assay Kit (Green) |
E-BC-K138-F |
|
Reactive Oxygen Species (ROS) Fluorometric Assay Kit (Red) |
E-BC-F005 |
|
Mitochondrial Superoxide Fluorometric Assay Kit |
E-BC-F008 |
|
MitoBright Green Probe Assay Kit |
E-CK-A401 |
|
MitoBright Red Probe Assay Kit |
E-CK-A402 |
|
MitoBright Deep Red Probe Assay Kit |
E-CK-A403 |
|
Cell Mitochondrial Extraction Assay Kit |
E-BC-E006 |
|
Animal Tissue Mitochondrial Extraction Assay Kit |
E-BC-E001 |
|
Fatty Acid Oxidation (FAO) Colorimetric Assay Kit |
E-BC-K784-M |
|
Mitochondrial Complex II Activity Assay Kit |
E-BC-K150-M |
02 Measuring cytotoxic function loss in exhausted OT-1 CD8+ T cells
The evaluation of cytotoxic function loss in exhausted OT-1 CD8+ T cells necessitates a multi-dimensional analytical strategy integrating phenotypic characterization, cytolytic granule release functional assays, and metabolic profiling. OT-1 T cells harbor a transgenic T cell receptor (TCR) that specifically recognizes the ovalbumin-derived peptide SIINFEKL, making them an ideal canonical model for elucidating the hierarchical deterioration of T cell effector functions under persistent antigen stimulation. Cytotoxic impairment in exhausted OT-1 T cells does not manifest as a complete ablation of cytolytic activity, but as a progressive functional deficit marked by compromised polyfunctionality, attenuated cellular degranulation, and dysregulated transcriptional programming driven by sustained TCR signaling. This progressive loss of cytotoxic competence is tightly coupled with metabolic dysfunction. Mitochondrial structural and functional defects, together with impaired glycolytic metabolism, collectively sustain and consolidate the T cell exhausted phenotype[10].
Phenotypic identification of exhausted OT-1 T cells relies on the persistent surface expression of multiple inhibitory receptors, including PD-1, TIM-3, LAG-3, and TIGIT (t cell exhaustion markers, which can be detected by PD-1 antibody, TIM-3 antibody, LAG-3 antibody and TIGIT antibody). These biomarkers reliably distinguish exhausted T cell populations from functional effector and memory T cell subsets. However, sole reliance on surface phenotypic markers is insufficient for quantifying cytotoxic functional impairment, given that the expression levels of these inhibitory receptors do not always correlate linearly with T cell cytolytic capacity. For this reason, functional validation assays are indispensable for directly assessing the ability of OT-1 T cells to eliminate target cells bearing cognate SIINFEKL antigens. Standard in vitro cytotoxicity assays co-culture OT-1 T cells with SIINFEKL-pulsed target cells, followed by the quantification of target cell lysis through flow cytometry-based viability dye staining or chromium-51 release detection. Even after normalizing for cell number, exhausted OT-1 T cells consistently exhibit markedly reduced target cell killing efficiency compared with acute effector OT-1 T cells in such assays[11].
The release of cytolytic granules containing perforin and granzymes constitutes the core functional basis of T cell cytotoxicity. Exhausted OT-1 T cells display substantially diminished expression of perforin and granzyme B, which directly impairs their capacity to trigger target cell apoptosis. CD107a (LAMP-1) degranulation assays, which measure surface CD107a exposure upon antigen restimulation, serve as a sensitive quantitative indicator of this granule release dysfunction. Following antigen stimulation, exhausted OT-1 T cells show severely blunted CD107a upregulation, indicating defective intracellular trafficking and plasma membrane fusion of cytolytic granules. Furthermore, this degranulation deficiency is aggravated by the immunosuppressive tumor microenvironment (TME), in which nutrient deprivation and the accumulation of inhibitory metabolites (e.g., lactate) robustly suppress cytolytic granule exocytosis[12,13].
Hierarchical impairment of cytokine production is a core hallmark of T cell exhaustion, occurring in parallel with the progressive decline of cytotoxic function. During OT-1 T cell exhaustion progression, interleukin-2 (IL-2) production is ablated at the early differentiation stage, followed by gradual reductions in tumor necrosis factor-alpha (TNF-α) and interferon-gamma (IFN-γ) secretion. While progenitor exhausted T cells (Tpex) retain low-level IFN-γ production, terminally exhausted T cells (Tex) exhibit profound, comprehensive deficits in the production of all three key cytokines. Combined intracellular cytokine staining and flow cytometry enables simultaneous detection of multiple cytokines and cytotoxic markers, thereby allowing comprehensive profiling of T cell functional impairment. Notably, IL-2 loss is particularly detrimental, as it disrupts the autocrine signaling required for T cell proliferation and survival, which further restricts the clonal expansion of cytotoxic OT-1 T cells[14,15].
Metabolic reprogramming acts as the fundamental driver of functional deficits in exhausted OT-1 T cells. Chronic TCR stimulation induces severe mitochondrial dysfunction, which is characterized by fragmented mitochondrial networks, reduced mitochondrial membrane potential, and compromised oxidative phosphorylation (OXPHOS). Such mitochondrial functional collapse severely limits intracellular adenosine triphosphate (ATP) availability, failing to support the energy-demanding processes of cytolytic granule synthesis and release. In addition, exhausted OT-1 T cells exhibit impaired glycolytic capacity. This defect is partially attributed to PD-1-mediated suppression of the PI3K-Akt-mTOR signaling pathway. The resulting metabolic insufficiency renders T cells incapable of meeting the high biosynthetic and bioenergetic demands of cytotoxic effector responses. Emerging evidence further demonstrates that mitochondrial dysfunction facilitates the transition from the Tpex to Tex state via HIF-1α-dependent glycolytic reprogramming, ultimately locking OT-1 T cells into a terminally dysfunctional exhausted state[16,17].
The TME further exacerbates OT-1 T cell cytotoxic dysfunction through metabolic competition and immunosuppressive metabolite accumulation. Tumor cells rely on robust glycolysis and produce abundant lactate, which accumulates within the TME and impairs T cell function via MCT11-mediated intracellular uptake. Lactate buildup disrupts intracellular pH homeostasis and key signaling cascades, thereby directly inhibiting the cytotoxic molecular machinery of OT-1 T cells. Moreover, TME hypoxia stabilizes HIF-1α expression, which redirects cellular metabolism toward unsustainable glycolysis and further suppresses mitochondrial respiratory function. These extrinsic microenvironmental stresses synergize with intrinsic T cell metabolic defects to form a potent immunosuppressive niche that severely compromises the cytotoxic potential of exhausted OT-1 T cells[18].
Transcriptional regulation and epigenetic remodeling also play indispensable roles in stabilizing cytotoxic functional loss in exhausted OT-1 T cells. The master exhaustion transcription factors TOX and NR4A are significantly upregulated in exhausted OT-1 T cells, where they drive sustained inhibitory receptor expression while transcriptionally repressing effector function-associated genes. These transcription factors trigger widespread epigenetic modifications, including histone remodeling and DNA methylation, which permanently stabilize the exhausted T cell phenotype. Importantly, such epigenetic scars persist even after metabolic improvement in the microenvironment, rendering cytotoxic dysfunction partially irreversible in the absence of combinatorial therapeutic intervention. A comprehensive understanding of these multi-layered regulatory mechanisms spanning phenotypic, functional, metabolic, transcriptional, and epigenetic levels is critical for developing targeted strategies to reverse T cell exhaustion, restore cytotoxic function, and improve the efficacy of cancer immunotherapy[19,20].
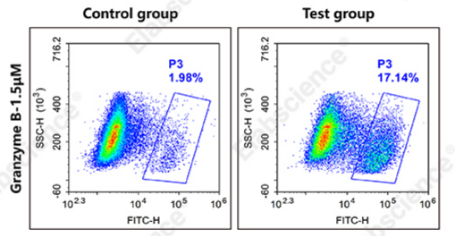
Fig. 2 Detection and analysis of activated granzyme B in Jurkat cells. Jurkat cells were cultured with (test group) or without (control group) 1×Cell Stimulation MIX for 5 h, followed by staining with Green-fluorescent human granzyme B substrate. Flow cytometry was used for subsequent analysis. (The data are provided by Elabscience.)
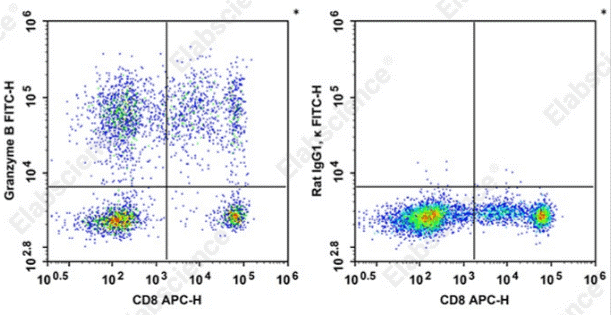
Fig. 3 Detection and analysis of Granzyme B in normal human peripheral blood cells. Normal human peripheral blood cells were stained with APC Anti-Human CD8a Antibody[OKT-8] and intracellular stained with FITC Anti-Human Granzyme B Antibody[QA18A28] (left) or FITC Rat IgG1, κ Isotype Control (right). Cells in the monocytes gate were used for analysis. (The data are provided by Elabscience.)
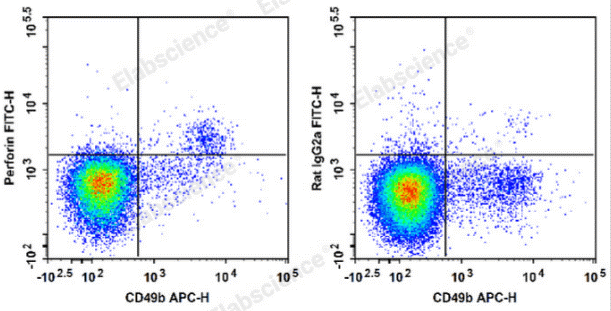
Fig. 4 Detection and analysis of perforin in C57BL/6 murine splenocytes. C57BL/6 murine splenocytes are stained with APC Anti-Mouse CD49b Antibody and FITC Anti-Mouse Perforin Antibody (Left). Splenocytes are stained with APC Anti-Mouse CD49b Antibody and FITC Rat IgG2a, κ Isotype Control (Right). (The data are provided by Elabscience.)
Elabscience® Quick Overview of Popular Products:
Table 2. Reagents for OT-1 CD8+ T cells cytotoxic function research
|
Product Name |
Cat. No. |
|
FITC Anti-Mouse Perforin Antibody[S16009A] |
E-AB-F1294C |
|
APC Anti-Mouse CD49b/pan-NK cells Antibody[DX5] |
E-AB-F1116E |
|
FITC Anti-Human Granzyme B Antibody[QA18A28] |
AN00961C |
|
APC Anti-Human CD8a Antibody[OKT-8] |
E-AB-F1110E |
|
Human Granzyme B Activity Detection Substrate for Flow Cytometry |
E-CK-A480 |
|
Purified Anti-Human/Mouse Granzyme B Antibody[QA16A02] |
AN009690P |
|
Purified Anti-Human Perforin Antibody[δG9] |
AN010970P |
|
Intracellular Fixation/Permeabilization Buffer Kit |
E-CK-A109 |
|
Enhanced ATP Chemiluminescence Assay Kit |
E-BC-F201 |
03 Comparing exhaustion signatures between OT-1 and polyclonal CD8+ T cells
Comparative analysis of exhaustion signatures between OT-1 transgenic CD8+ T cells and polyclonal endogenous CD8+ T cells reveals conserved core molecular programs and distinct contextual divergences driven by antigen specificity, precursor frequency, and microenvironmental constraints. OT-1 cells serve as a well-controlled experimental model for exploring the mechanistic drivers of T cell exhaustion under chronic antigen stimulation. In contrast, polyclonal T cell populations display markedly higher heterogeneity in differentiation trajectories, which reflects the diverse TCR affinity landscapes and clonal competitive dynamics of physiological in vivo immune responses. The core transcriptional and epigenetic machinery governing T cell exhaustion includes the upregulation of multiple inhibitory receptors and the induction of master regulatory transcription factors such as TOX and NR4A. This core machinery is largely conserved in both OT-1 and polyclonal systems. Nonetheless, the magnitude and temporal kinetics of these molecular alterations vary significantly between the two models[21].
Sustained high expression of multiple inhibitory receptors, predominantly PD-1, TIM-3, LAG-3, and TIGIT, constitutes the fundamental phenotypic hallmark of T cell exhaustion. In canonical OT-1 experimental systems, including chronic lymphocytic choriomeningitis virus (LCMV) infection and ovalbumin-expressing tumor models, this exhausted phenotype develops rapidly. The rapid onset of exhaustion is attributed to the uniform high-affinity recognition of the cognate SIINFEKL antigen by monoclonal OT-1 TCRs. Conversely, polyclonal endogenous CD8+ T cells exhibit a broad spectrum of inhibitory receptor co-expression patterns modulated by variable TCR affinities and epitope immunodominance within the endogenous repertoire. High-affinity clones in polyclonal populations typically develop deep exhaustion that phenotypically and functionally recapitulates the exhaustion observed in OT-1 cells. In comparison, low-affinity polyclonal clones often retain partial effector functionality or enter alternative dysfunctional states that do not fully match the canonical exhausted transcriptomic profile. Such clonal heterogeneity in polyclonal responses underscores the critical roles of antigen load and antigen quality in shaping the exhaustion landscape. These variables are tightly standardized and controlled in simplified OT-1 experimental systems but remain highly dynamic during natural infection and tumor progression[22].
At the transcriptional level, both OT-1 and polyclonal exhausted CD8+ T cells rely on conserved core regulatory circuits, with TOX and NR4A acting as central drivers of the exhausted cell state. Persistent TCR signaling directly induces TOX expression, which is indispensable for the establishment of the exhausted phenotype. TOX concurrently promotes the expression of multiple inhibitory receptors and represses genes associated with effector functions. In OT-1 cells, TOX expression levels strongly correlate with exhaustion severity, particularly in terminally differentiated exhausted subsets. Polyclonal T cells retain TOX-dependent regulatory programming, yet the correlation between TOX abundance and functional impairment is more nuanced in endogenous populations. Certain polyfunctional memory-like T cell subsets maintain low basal TOX expression without developing severe dysfunction. Furthermore, the transcription factor TCF1, encoded by the TCF7 gene, marks the progenitor exhausted T cell (Tpex) subset in both systems. These Tpex cells possess self-renewal capacity and remain responsive to immune checkpoint blockade therapy. Despite conserved TCF1-mediated progenitor programming, the proportion of TCF1+ progenitor cells within the total exhausted pool differs substantially between OT-1 and polyclonal responses. This variation is modulated by antigen exposure duration and local inflammatory cytokine availability in the microenvironment[23].
Epigenetic remodeling is a key deterministic mechanism that stabilizes the exhausted phenotype in both OT-1 and polyclonal CD8+ T cells. Widespread chromatin modifications permanently lock T cells in a dysfunctional state. Exhausted CD8+ T cells exhibit unique chromatin accessibility profiles that distinguish them from canonical effector and memory T cells. Specifically, chromatin regions linked to effector functions display reduced transcriptional accessibility, whereas loci encoding inhibitory receptors remain constitutively open and transcriptionally active. These stable epigenetic scars form at the early stage of chronic antigen stimulation and are largely irreversible, thereby limiting functional recovery even after antigen clearance and stimulation resolution. Uniform antigen exposure in OT-1 models drives highly synchronized epigenetic reprogramming across the entire T cell population. By contrast, polyclonal T cell responses show substantial chromatin remodeling variability resulting from the asynchronous activation and differentiation of individual clonal populations. Recent multi-omic profiling studies confirm the conservation of core exhaustion-associated epigenetic landscapes across different T cell models. Meanwhile, distinct regulatory elements exhibit differential chromatin accessibility in polyclonal T cell populations, providing potential targets for the selective rejuvenation of high-affinity exhausted T cell clones[24].
Metabolic reprogramming is another conserved functional hallmark of T cell exhaustion, characterized by pervasive mitochondrial dysfunction and impaired glycolytic capacity. Both OT-1 and polyclonal exhausted CD8+ T cells exhibit reduced mitochondrial membrane potential, fragmented mitochondrial networks, and compromised oxidative phosphorylation. Persistent inhibitory receptor signaling, particularly PD-1-mediated pathway suppression, drives this metabolic insufficiency. PD-1 signaling inhibits the downstream PI3K-Akt-mTOR axis and restricts cellular glucose uptake, thus disrupting core bioenergetic processes. In OT-1 cells, the severity of metabolic defects is tightly coupled with impaired cytotoxic activity and cytokine secretion, with terminally exhausted subsets exhibiting the most severe mitochondrial damage. Polyclonal T cell populations present similar core metabolic impairments, yet the degree of dysfunction varies considerably among individual clones. A subset of polyclonal clones retains partial metabolic flexibility and residual effector potential under chronic stimulation. The tumor microenvironment further exacerbates these metabolic deficits in both T cell systems through nutrient competition and the accumulation of immunosuppressive metabolites. Lactate, a key inhibitory metabolite, impairs T cell function via MCT11-dependent intracellular uptake and ultimately suppresses T cell cytotoxicity and effector function[25,26].
Functional assays reveal divergent patterns of functional deterioration despite conserved exhaustion features in OT-1 and polyclonal exhausted CD8+ T cells. In OT-1 monoclonal models, IL-2 production is lost at the earliest stage of exhaustion progression, followed by gradual reductions in TNF-α and IFN-γ secretion. This sequential functional deterioration ultimately leads to severe defects in granule degranulation and target cell killing. In contrast, polyclonal endogenous T cell responses exhibit a more graded and heterogeneous pattern of functional loss. Multiple polyclonal clones sustain robust IFN-γ production even with high PD-1 expression and persistent exhaustion. This functional discrepancy stems from variable TCR affinity and clonal competition within the endogenous T cell repertoire. High-affinity polyclonal clones undergo deep terminal exhaustion similar to OT-1 cells, while low-affinity clones maintain a partially functional, less impaired state. Furthermore, CD4+ T cell helper signals effectively modulate the exhaustion trajectory of polyclonal CD8+ T cells. This physiological regulatory factor is often eliminated or strictly controlled in simplified OT-1 experimental systems, forming a key contextual gap between preclinical models and in vivo physiological responses[27,28].
In summary, OT-1 transgenic and polyclonal endogenous CD8+ T cells share core molecular features of T cell exhaustion, including inhibitory receptor upregulation, TOX-dominated transcriptional programming, stable epigenetic locking, and pervasive metabolic dysfunction. However, the two systems differ greatly in the heterogeneity, magnitude, and kinetics of exhaustion progression. OT-1 models recapitulate synchronized and simplified T cell exhaustion induced by uniform high-affinity antigen stimulation. In comparison, polyclonal T cell responses reflect the complex physiological interplay of diverse TCR affinities, clonal competition, and dynamic microenvironmental pressure. A comprehensive understanding of these conserved and divergent characteristics is essential for translating OT-1 preclinical findings to clinical applications. The inherent diversity of the human endogenous T cell repertoire critically determines immune surveillance efficiency and patient responses to cancer immunotherapy.
_.png)
Fig. 5 Analysis of IL-17A expression in human peripheral blood mononuclear cells (PBMCs). PBMCs were stimulated with Cell Stimulation MIX combined with Protein Transport Inhibitor MIX for 5 h. Subsequently, cells were collected, fixed and permeabilized, and immunolabeled using PerCP/Cyanine5.5 anti-human CD3, Elab Fluor® 488 anti-human CD4 and PE anti-human IL-17A antibodies. Flow cytometry was applied to quantify the frequency and IL-17A secretory function of Th17 cells, which were phenotypically defined as CD3+CD4+IL-17A+cells. (The data are provided by Elabscience.)
Elabscience® Quick Overview of Popular Products:
Table 3. Reagents for NK Cell Research
|
Product Name |
Cat. No. |
|
EasySort™ Mouse NK Cell Isolation Kit |
MIM005N |
|
EasySort™ Human Naïve CD8+T Cell Isolation Kit |
MIH008N |
|
EasySort™ Mouse Naïve CD8+T Cell Isolation Kit |
MIM008N |
|
EasySort™ Human CD8+ T Cell Isolation Kit |
MIH003N |
|
EasySort™ Mouse CD8+T Cell Isolation Kit |
MIM003N |
|
Mouse Th17 Flow Cytometry Staining Kit |
XJM002 |
|
Mouse Th1/Th2 Flow Cytometry Staining Kit |
XJM001 |
|
Human Th1/Th2 Flow Cytometry Staining Kit |
XJH001 |
|
Human Th17 Flow Cytometry Staining Kit |
XJH002 |
|
Anti-Human CD45-FITC/CD3-PE/Cyanine5/CD56/NCAM-PE/CD19-PE/Elab Fluor® 594 Cocktail |
E-AB-FC0028 |
|
Anti-Human CD19-FITC/CD56-PE/CD3-PE/Cyanine7/CD45-PerCP Cocktail |
E-AB-FC0011 |
|
EasyStain™ Human Fc Receptor Blocking Solution |
E-CK-A171 |
|
FITC Anti-Human/Monkey CD16 Antibody[3G8] |
E-AB-F1236C |
|
PE Anti-Human HLA-DR Antibody[L243] |
E-AB-F1111D |
|
PerCP/Cyanine5.5 Anti-Human CD3 Antibody[OKT-3] |
E-AB-F1001J |
|
APC Anti-Human/Monkey CD56/NCAM Antibody[B-A19] |
E-AB-F1305E |
04 Monitoring PD-1, TIM-3, and LAG-3 expression during OT-1 T cell expansion
Monitoring the expression of inhibitory receptors including PD-1, TIM-3, and LAG-3 during OT-1 CD8+ T cell expansion provides critical insights into the temporal dynamics of T cell exhaustion under chronic antigen stimulation. The OT-1 transgenic model generates T cell receptors that specifically recognize the ovalbumin-derived peptide SIINFEKL. This model enables precise tracking of surface inhibitory markers whose dynamic expression patterns correlate with progressive functional decline and metabolic reprogramming. The co-expression profiles of these inhibitory receptors are not static. Instead, they evolve continuously to reflect stepwise T cell differentiation from early effector states to progenitor exhausted (Tpex) subsets and ultimately terminally exhausted (Tex) subsets. Elucidating this dynamic trajectory is essential for clarifying the molecular mechanisms driving persistent T cell dysfunction. It also helps define optimal therapeutic windows during which checkpoint blockade immunotherapy achieves maximal efficacy[29].
PD-1 upregulation represents the earliest and most consistent molecular signature of sustained chronic antigen exposure in expanding OT-1 T cells. Acute antigen stimulation induces only transient PD-1 expression that closely accompanies T cell activation. In contrast, chronic antigen stimulation drives persistent and high-level PD-1 expression throughout T cell expansion. Continuous TCR signaling sustains this basal PD-1 expression, and inflammatory cytokines within the tissue microenvironment further amplify this phenotype. In OT-1 experimental systems, PD-1 expression intensity strongly correlates with exhaustion severity, particularly in terminally differentiated subsets with diminished proliferative capacity. However, PD-1 expression alone cannot reliably define exhausted T cell status, given that activated functional effector T cells also transiently express PD-1. Accordingly, PD-1 detection must be integrated with additional inhibitory receptor profiling to accurately quantify the degree of T cell functional impairment[30].
TIM-3 and LAG-3 expression occurs subsequent to PD-1 induction, marking a more advanced exhaustion stage with aggravated functional defects. OT-1 T cells co-expressing PD-1 and TIM-3 exhibit significantly impaired cytokine production and cytotoxic activity relative to cells expressing PD-1 alone. This hierarchical acquisition of multiple inhibitory receptors reflects progressive epigenetic fixation of the exhausted phenotype, as chromatin accessibility at inhibitory receptor loci gradually increases during persistent stimulation. LAG-3 upregulation further correlates with severe metabolic insufficiency, particularly mitochondrial dysfunction that restricts energy supply for core T cell effector functions. The simultaneous expression of PD-1, TIM-3, and LAG-3 defines a deeply exhausted T cell population that is largely unresponsive to single-agent checkpoint blockade therapy (which can be detected by PD-1 antibody, TIM-3 antibody, LAG-3 antibody and TIGIT antibody)[29,30].
Inhibitory receptor expression patterns directly reflect subset heterogeneity within expanding OT-1 T cell populations. TCF1-expressing progenitor exhausted T cells (Tpex) maintain high PD-1 levels but exhibit relatively low TIM-3 and LAG-3 expression. This unique phenotypic profile preserves Tpex self-renewal capacity and responsiveness to immune checkpoint inhibitors, enabling these cells to serve as a renewable reservoir for sustaining the overall T cell pool. In comparison, terminally exhausted T cells (Tex) lack TCF1 expression and display robust co-expression of all three inhibitory receptors. This profile defines a fixed terminal differentiation state with limited therapeutic reversibility. Monitoring the dynamic ratio of Tpex to Tex subsets during T cell expansion therefore provides a reliable metric for evaluating the overall functional quality of OT-1 T cell immune responses[31].
Metabolic reprogramming serves as a central upstream driver of inhibitory receptor upregulation during OT-1 T cell exhaustion. Chronic TCR signaling suppresses the PI3K-Akt-mTOR pathway, which induces defective glycolysis and profound mitochondrial dysfunction. The resulting cellular metabolic insufficiency activates key downstream transcription factors including HIF-1α and TOX. These transcription factors directly initiate and sustain the expression of PD-1, TIM-3, and LAG-3. Specifically, mitochondrial stress under persistent antigen stimulation and hypoxic conditions rapidly accelerates inhibitory receptor upregulation, establishing a direct mechanistic link between metabolic dysfunction and phenotypic exhaustion. Furthermore, tumor microenvironment-derived lactate accumulates intracellularly via MCT11-mediated transport. Lactate accumulation further stabilizes inhibitory receptor expression and consolidates the irreversible dysfunctional T cell state[32].
Epigenetic remodeling acts as the fundamental mechanism that stabilizes persistent inhibitory receptor expression in exhausted OT-1 T cells. Exhausted T cells exhibit distinct chromatin accessibility signatures. Effector function-associated loci remain transcriptionally repressed with condensed chromatin, whereas inhibitory receptor loci maintain constitutively open and transcriptionally active chromatin states. These epigenetic alterations are established at the early phase of chronic stimulation and remain largely irreversible. Such stable epigenetic scars sustain inhibitory receptor expression even after antigen clearance. The transcription factor TOX is indispensable for this process, as it orchestrates widespread epigenetic remodeling to lock T cells into a terminal exhausted lineage. Consequently, profiling inhibitory receptor expression not only reflects real-time T cell functional status but also indicates the extent of epigenetic commitment to the exhausted phenotype[33].
In conclusion, dynamic profiling of PD-1, TIM-3, and LAG-3 expression during OT-1 T cell expansion reveals a sophisticated regulatory network interconnected by antigen stimulation, metabolic reprogramming, and epigenetic modification. The hierarchical acquisition of these inhibitory receptors precisely demarcates the transition from progenitor to terminal exhaustion, which directly determines T cell therapeutic responsiveness. Understanding these spatiotemporal exhaustion dynamics is critical for developing advanced T cell rejuvenation strategies. The combination of checkpoint blockade with metabolic targeted interventions can effectively reverse T cell dysfunction and restore effector capacity. The OT-1 model continues to provide robust and reproducible mechanistic insights into T cell exhaustion, emphasizing the necessity of multi-parameter phenotypic and functional analysis for accurate exhaustion evaluation in cancer immunology research.
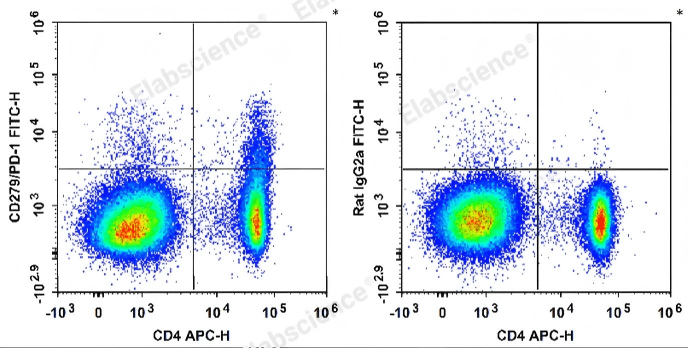
Fig. 6 Detection and analysis of PD-1 expression in splenocytes from C57BL/6 mice. Splenocytes were stained with APC-conjugated anti-mouse CD4 antibody and FITC-conjugated anti-mouse CD279/PD-1 antibody (left). For isotype control, cells were labeled with APC-conjugated anti-mouse CD4 antibody and FITC-conjugated rat IgG2a, κ isotype control (right). (The data are provided by Elabscience.)
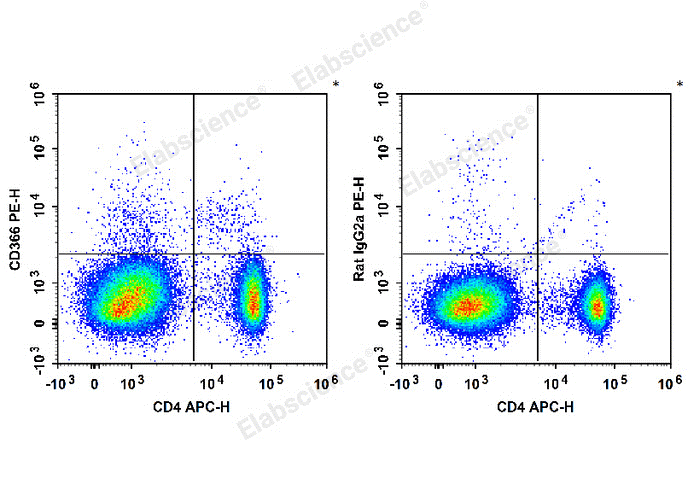
Fig. 7 Detection and analysis of TIM3/CD366 in splenocytes from C57BL/6 mice. C57BL/6 murine splenocytes are stained with APC Anti-Mouse CD4 Antibody and PE Anti-Mouse CD366/Tim-3 Antibody (Left). For isotype control, cells were labeled with APC Anti-Mouse CD4 Antibody and PE Rat IgG2a, κ Isotype Control (Right). (The data are provided by Elabscience.)
Elabscience® Quick Overview of Popular Products:
Table 4. Reagents for T cell exhaustion research
|
Product Name |
Cat. No. |
|
Purified Anti-Human CD366 Antibody[F38-2E2], Functional Grade |
AN009710 |
|
PE Anti-Human CD279/PD-1 Antibody[EH12.2H7] |
E-AB-F1229D |
|
Elab Fluor® Violet 450 Anti-Human CD45 Antibody[HI30] |
E-AB-F1137Q |
|
Elab Fluor® Red 780 Anti-Human CD3 Antibody[OKT-3] |
E-AB-F1001S |
|
FITC Anti-Human/Monkey CD4 Antibody[SK3] |
E-AB-F1352C |
|
PerCP/Cyanine5.5 Anti-Human CD8 Antibody[UCHT-4] |
AN00427J |
|
PE Anti-Human CD25 Antibody[BC96] |
E-AB-F1194D |
|
APC Anti-Human CD127/IL-7RA Antibody[A019D5] |
E-AB-F1152E |
|
FITC Anti-Mouse CD279/PD-1 Antibody[29F.1A12] |
E-AB-F1131C |
|
APC Anti-Mouse CD4 Antibody[GK1.5] |
E-AB-F1097E |
|
PE Anti-Mouse CD366/Tim-3 Antibody[RMT3-23] |
E-AB-F1192D |
|
10×ACK Lysis Buffer |
E-CK-A105 |
|
10× RBC Lysis/Fixation Solution |
E-CK-A106 |
05 Cytokine supplementation strategies for functional maintenance of OT-1 CD8+ T cells and current research gaps
Current literature lacks a systematic summary of cytokine supplementation strategies optimized to preserve the functional integrity of OT-1 CD8+ T cells (OT-1 CD8 T cells). Drawing on preclinical findings from published OT-1 studies, this section consolidates commonly used cytokine regimens and their corresponding experimental outcomes.
5.1 In Vitro Culture and Activation Strategies
For the in vitro activation and expansion of OT-1 CD8+ T cells (OT-1 CD8 T cells), interleukin-2 (IL-2) supplementation at 10 ng/mL is the most established and rigorously validated cytokine strategy. IL-2 binds to IL-2 receptors on activated T cells to support continuous proliferation and survival, while sustaining core effector functions including interferon-γ (IFN-γ) secretion and cytotoxicity. As such, IL-2 supplementation serves as an indispensable baseline condition for maintaining the fundamental functionality of cultured OT-1 cells (T cell culture)[34].
Nevertheless, no consistent consensus has been established regarding the necessity of supplementing additional cytokines during routine in vitro OT-1 culture. Several exploratory optimization strategies have been reported for experimental designs that target specific T cell functional phenotypes.
Combining reduced IL-2 dosage with low-dose interleukin-15 (IL-15) is a feasible approach to promote the generation of long-lived memory T cells. However, this combinatorial regimen lacks large-scale validation specifically in standardized OT-1 experimental models[34].
Interleukin-27 (IL-27) modulates the differentiation of pathogenic CD8+ T cells and enhances their effector functions under specific contexts. These effects are highly condition-dependent and have been primarily characterized in autoimmune disease models. Thus, IL-27 is not suitable as a conventional strategy for maintaining OT-1 CD8+ T cell functionality[35].
5.2 In Vivo Application and Immunotherapeutic Combination Strategies
In in vivo experimental systems, especially adoptive OT-1 T cell therapy, most existing studies predominantly focus on endogenous cytokine regulation. In contrast, research exploring direct exogenous cytokine supplementation remains limited. Key experimental conclusions are summarized as follows.
(1) Interleukin-12 (IL-12): Accumulated evidence indicates that endogenous IL-12 is dispensable for the initial expansion, differentiation, and effector function acquisition of vaccine-stimulated OT-1 CD8+ T cells (OT-1 CD8 T cells). Deficiency in endogenous IL-12 does not alter the proliferation, viability, interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) secretion, or degranulation capacity of OT-1 cells. Although mRNA-encoded IL-12 is widely applied as an adjuvant to amplify systemic CD8+ T cell immune responses, no definitive evidence verifies that exogenous IL-12 supplementation specifically enhances the functional performance of OT-1 CD8+ T cells (OT-1 CD8 T cells)[36].
(2) Combination with immune checkpoint modulation: Within the immunosuppressive tumor microenvironment (TME), combined treatment with programmed death-1 (PD-1) inhibitors and 4-1BB agonists effectively recruits more tumor-specific OT-1 CD8+ T cells into tumor tissues. This combinatorial strategy remodels the immunosuppressive TME and indirectly sustains T cell effector function, serving as a prevalent in vivo approach to boost OT-1 cell activity. Notably, such functional improvement depends on microenvironmental remodeling rather than direct cytokine supplementation[37].
5.3 Current Research Gaps and Prospective Directions
Most existing OT-1 studies utilize this transgenic model merely to validate the regulatory mechanisms of target molecules. Direct head-to-head comparisons of diverse cytokine combinations and optimized supplementation regimens for OT-1 functional maintenance remain underexplored. To date, the most reliable and widely adopted baseline protocol for standard in vitro OT-1 experiments combines CD3/CD28-mediated antigenic activation with conventional IL-2 supplementation[38].
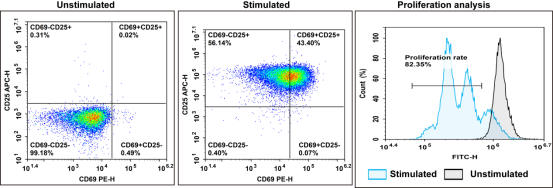
Fig. 8 In vitro sorting, activation and culture of CD8+ T cells. Fresh human PBMCs were isolated, and CD8+ T cells were purified via the EasySort™ Human CD8⁺ T Cell Isolation Kit. Cells were activated with CD3/CD28 activation beads and cultured for 3 days. The expression of CFSE, CD69 and CD25 was analyzed by flow cytometry. (The data are provided by Elabscience.)
Table 5. Reagents for OT-1 CD8+ T cell activation
|
Product Name |
Cat. No. |
|
Human CD3/CD28 T Cell Activation Beads |
MIH001A |
|
Mouse CD3/CD28 T Cell Activation Beads |
MIM001A |
|
Cell Stimulation and Protein Transport Inhibitor Kit |
E-CK-A091 |
|
EasyStain™ Human Fc Receptor Blocking Solution |
E-CK-A171 |
|
CFSE Cell Division Tracker Kit |
E-CK-A345 |
|
Intracellular Fixation/Permeabilization Buffer Kit |
E-CK-A109 |
|
APC Anti-Human CD69 [FN50] |
E-AB-F1138E |
|
PE Anti-Human CD25 Antibody[BC96] |
E-AB-F1194D |
|
FITC Anti-Human HLA-DR [L243] |
E-AB-F1111C |
|
PerCP/Cyanine5.5 Anti-Human CD3 [UCHT1] |
E-AB-F1230J |
06 APC selection in OT-1 co-culture models: splenocytes, BMDCs, or artificial APCs
In OT-1 CD8+ T cell co-culture assays, the selection of antigen-presenting cells (APCs) should be tailored to specific experimental objectives. In addition to the conventional CD3/CD28 T cell activation strategy for in vitro T cell stimulation, various APC platforms support physiological antigen-specific activation of OT-1 T cells with distinct functional characteristics. Splenocytes, bone marrow-derived dendritic cells (BMDCs), and artificial antigen-presenting cells (aAPCs) all exhibit unique strengths and limitations in stimulating OT-1 T cells, which are systematically summarized in the following sections.
6.1 Splenocytes
Splenocytes are crude yet easily accessible APC populations directly isolated from murine spleens, consisting of a small proportion of endogenous dendritic cells and other professional APCs[39].
(1) Advantages: Splenocytes can be prepared through an extremely simple and rapid procedure without the need for complex in vitro differentiation induction, thus enabling low-cost experimental operations. Moreover, they preserve the native mixed immune microenvironment of the spleen, making them well-suited for preliminary screening and rapid functional validation experiments. In OT-1 experimental systems, splenic myeloid subsets have been proven to efficiently present ovalbumin (OVA) antigens and robustly drive OT-1 T cell proliferation.
(2) Disadvantages: As a highly heterogeneous mixed cell population, splenocytes contain only a low fraction of professional APCs, which results in unstable antigen stimulation efficiency. Furthermore, the quantity and maturation status of functional APCs cannot be precisely controlled, greatly limiting their application in standardized quantitative experiments.
(3) Applicable scenarios: Preliminary small-scale exploratory experiments, simple validation of antigen presentation function, and research that requires the preservation of endogenous immune cell composition and the native splenic immune microenvironment.
6.2 Bone marrow-derived dendritic cells (BMDCs)
BMDCs are professional dendritic cells derived from the in vitro induced differentiation of murine bone marrow progenitor cells and are widely recognized as the gold-standard APCs for in vitro OT-1 T cell co-culture experiments[40].
(1) Advantages: BMDCs possess high cellular purity and a high proportion of professional APCs, and acquire potent antigen-presenting capacity upon stimulation with maturation inducers such as lipopolysaccharide (LPS). Since the quantity and maturation degree of BMDCs can be artificially modulated, experiments using BMDCs exhibit excellent repeatability. As the most commonly adopted APCs in OT-1 co-culture studies, BMDCs can efficiently process and present antigen peptides to activate OT-1 CD8+ T cells (OT-1 CD8 T cells) and induce interferon-γ (IFN-γ) secretion.
(2) Disadvantages: BMDC preparation requires a complex and time-consuming in vitro differentiation process that lasts 7–10 days with granulocyte-macrophage colony-stimulating factor (GM-CSF) supplementation. This cumbersome procedure leads to elevated experimental costs and inevitable batch-to-batch variability in differentiation efficiency among individual mouse donors.
(3) Applicable scenarios: Standardized quantitative functional assays, mechanistic research on antigen presentation, and in vitro OT-1 T cell activation for subsequent in vivo adoptive transfer experiments.
6.3 Artificial APCs (aAPCs, artificial antigen presenting cells)
aAPCs are engineered nano- or microparticle-based synthetic APCs that reconstitute the core signaling cues essential for robust T cell activation[41,42].
(1) Advantages: aAPCs feature fully defined and controllable components, thereby ensuring superior experimental reproducibility. The surface density and categories of co-stimulatory molecules, as well as loaded cytokines, can be precisely customized to meet specific research requirements. In addition, magnetic aAPCs support antigen-specific enrichment and large-scale expansion of OT-1 T cells, serving as a convenient tool for high-yield OT-1 T cell preparation.
(2) Disadvantages: Unlike natural APCs, aAPCs are devoid of endogenous antigen processing and cross-presentation machineries and can only present predefined synthetic antigen peptides. Meanwhile, the fabrication or commercial purchase of aAPCs imposes high experimental costs. Accumulated studies have demonstrated that the functional performance of aAPCs is highly dependent on their particle size, ligand density, and other physicochemical properties, which necessitates systematic optimization for different experimental settings.
(3) Applicable scenarios: Mechanistic studies requiring precise modulation of T cell activation signaling, large-scale in vitro expansion of antigen-specific OT-1 T cells, and enrichment of rare OT-1 T cell subpopulations.
Table 6. Comparison of three APCs in OT-1 co-culture models
|
Comparison dimension |
Splenocytes |
BMDCs |
Artificial APCs |
|
Preparation difficulty |
Very low (direct isolation) |
High (7-10 days induction) |
Medium (customization/engineering) |
|
Antigen presentation capacity |
Low (low proportion of professional APC) |
High (professional DC) |
High (controllable signal strength) |
|
Repeatability |
Poor |
Medium |
Excellent |
|
Cost |
Low |
Medium |
High |
|
Can simulate endogenous antigen processing |
Yes |
Yes |
No (only exogenous peptides) |
For most conventional assays focusing on OT-1 T cell activation and functional validation, BMDCs remain the most preferable option by virtue of their balanced antigen activation efficiency and superior experimental reproducibility. For experiments prioritizing rapid preparation and cost reduction, splenocytes serve as a reliable and feasible alternative. In contrast, artificial APCs (artificial antigen presenting cells) are uniquely suited for customized mechanistic investigations and large-scale in vitro expansion of OT-1 T cells.
%20maturation_.png)
Fig. 9 Flow cytometric analysis of dendritic cell (DC) maturation. Bone marrow-derived DCs from C57BL/6 mice were exposed to maturation stimuli, which significantly increased the expression of MHC II, CD80, CD40 and CD86. (The data are provided by Elabscience.)
_.png)
Fig. 10 Interaction between T cells and bone marrow-derived dendritic cells (BMDCs). CD8+ T cells were isolated from the splenocytes of C57BL/6 mice using EasySort™ Mouse CD8+T Cell Isolation Kit (cat. no. MIM003N). The purity of the isolated CD8+ T cells was verified by flow cytometry. Following CFSE labeling, CD8+ T cells were co‑cultured with mature BMDCs for 72 h in vitro, and the proliferation of CD8+ T cells was subsequently assessed. (The data are provided by Elabscience.)
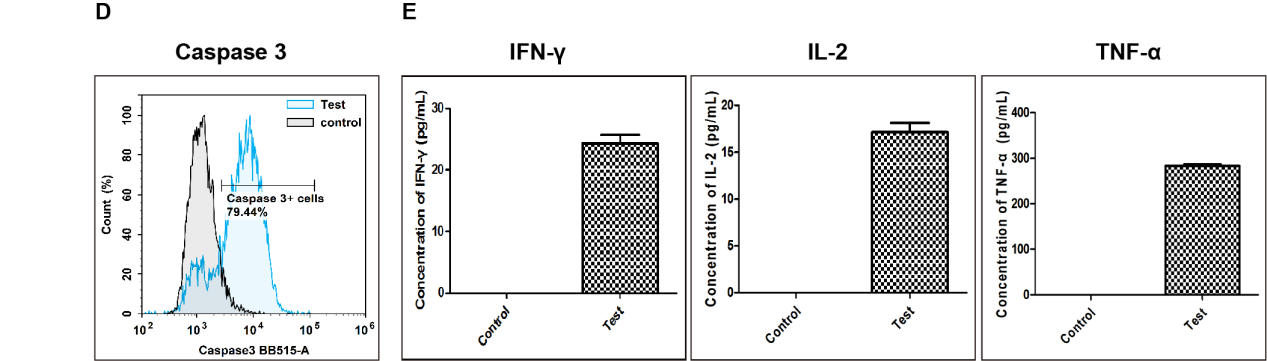
Fig. 11 Detection of apoptosis in target cells killed by CD8+ T cells. (D) After co‑culture of activated T cells with RAW264.7 target cells at a ratio of 1:10 for 24 h, caspase‑3 activity in target cells was measured by flow cytometry. Compared with the Control group (target cells cultured without activated T cells), the proportion of RAW264.7 cells with activated caspase‑3 increased to 79.44% in the co‑culture group (Test). (E) Cytokine levels in cell culture supernatants were measured by ELISA. Compared with the Control group, the levels of IFN‑γ, IL‑2, and TNF‑α were markedly increased in the co‑culture group (Test). (The data are provided by Elabscience.)
Elabscience® Quick Overview of Popular Products:
Table 7. Reagents for DC-T cell co-culture
|
Product Name |
Cat. No. |
|
EasySort™ Mouse CD8+T Cell Isolation Kit |
MIM003N |
|
EasySort™ Mouse CD3+T Cell Isolation Kit |
MIM001N |
|
EasySort™ Mouse CD4+T Cell Isolation Kit |
MIM002N |
|
EasySort™ Human CD3+T Cell Isolation Kit |
MIH001N |
|
EasySort™ Human CD4+ T Cell Isolation Kit |
MIH002N |
|
EasySort™ Human CD8+ T Cell Isolation Kit |
MIH003N |
|
EasySort™ Human Naïve Pan T Cell Isolation Kit |
MIH006N |
|
EasySort™ Human Naïve CD4+T Cell Isolation Kit |
MIH007N |
|
EasySort™ Human Naïve CD8+T Cell Isolation Kit |
MIH008N |
|
EasySort™ Mouse Pan-Naïve T Cell Isolation Kit |
MIM006N |
|
EasySort™ Mouse Naïve CD4+T Cell Isolation Kit |
MIM007N |
|
EasySort™ Mouse Naïve CD8+T Cell Isolation Kit |
MIM008N |
|
EasySort™-5 Magnet |
EC001 |
|
Mouse Bone Marrow-derived Dendritic Cells (BMDC) Induction and Identification Kit |
XJM003 |
|
FITC Anti-Mouse CD3 Antibody[17A2] |
E-AB-F1013C |
|
Elab Fluor® Violet 450 Anti-Mouse CD8a Antibody[53-6.7] |
E-AB-F1104Q |
|
Caspase 3/7 Activity Detection Substrate for Flow Cytometry |
E-CK-A483 |
|
Caspase 3/7 and Annexin V Double Staining Apoptosis Kit |
E-CK-A831 |
|
Caspase 1 Activity Detection Substrate for Flow Cytometry |
E-CK-A481 |
|
Reactive Oxygen Species (ROS) Fluorometric Assay Kit (Red) |
E-BC-F005 |
|
Lactate Dehydrogenase (LDH) Cytotoxicity Colorimetric Assay Kit |
E-BC-K771-M |
|
CellaQuant™ Mouse TNF-α (Tumor Necrosis Factor Alpha) ELISA Kit |
CQM002 |
|
CellaQuant™ Mouse IL-2 (Interleukin 2) ELISA Kit |
CQM006 |
|
CellaQuant™ Mouse IFN-γ (Interferon Gamma) ELISA Kit |
CQM005 |
Table.8 Multicolor Panel for Flow Cytometric Analysis of OT-1 CD+T Cells
|
Marker |
Clone |
Fluorochrome |
Cat. No. |
Species Reactivity |
|
CD45 |
30-F11 |
PerCP/Cyanine5.5 |
E-AB-F1136J |
Mouse |
|
CD3 |
17A2 |
FITC |
E-AB-F1013C |
Mouse |
|
CD4 |
GK1.5 |
PE |
E-AB-F1097D |
Mouse |
|
CD8a |
53-6.7 |
APC |
E-AB-F1104E |
Mouse |
|
CD45 |
HI30 |
Elab Fluor® Violet 450 |
E-AB-F1137Q |
Human |
|
CD3 |
OKT-3 |
APC |
E-AB-F1001E |
Human |
|
TCR γ/δ |
B1 |
FITC |
E-AB-F1145C |
Human |
|
CD45 |
HI30 |
Elab Fluor® Violet 450 |
E-AB-F1137Q |
Human |
|
CD3 |
UCHT1 |
Elab Fluor® Red 780 |
E-AB-F1230S |
Human |
|
CD4 |
SK3 |
FITC |
E-AB-F1352C |
Human |
|
CD8 |
OKT-8 |
PerCP/Cyanine5.5 |
E-AB-F1110J |
Human |
|
CD25 |
BC96 |
PE |
E-AB-F1194D |
Human |
|
CD127 |
A019D5 |
APC |
E-AB-F1152E |
Human |
References:
[1] GILES J R, NGIOW S F, MANNE S, et al. Shared and distinct biological circuits in effector, memory and exhausted CD8+ T cells revealed by temporal single-cell transcriptomics and epigenetics[J]. Nature Immunology, 2022, 23(11): 1600-1613.
[2] CHOW A, PERICA K, KLEBANOFF C A, et al. Clinical implications of T cell exhaustion for cancer immunotherapy[J]. Nature Reviews Clinical Oncology, 2022, 19(12): 775-790.
[3] SUN Q, DONG C. Regulators of CD8+ T cell exhaustion[J]. Nature Reviews Immunology, 2025, 26(2): 129-151.
[4] UTZSCHNEIDER D T, GABRIEL S S, CHISANGA D, et al. Early precursor T cells establish and propagate T cell exhaustion in chronic infection[J]. Nature Immunology, 2020, 21(10): 1256-1266.
[5] BAESSLER A, VIGNALI D A A. T Cell Exhaustion[J]. Annual Review of Immunology, 2024, 42(1): 179-206.
[6] WU H, ZHAO X, HOCHREIN S M, et al. Mitochondrial dysfunction promotes the transition of precursor to terminally exhausted T cells through HIF-1α-mediated glycolytic reprogramming[J]. Nature Communications, 2023, 14(1). http://dx.doi.org/10.1038/s41467-023-42634-3. DOI:10.1038/s41467-023-42634-3.
[7] QUEZADA L K, JIN W, LIU Y C, et al. Early transcriptional and epigenetic divergence of CD8+ T cells responding to acute versus chronic infection[J]. PLOS Biology, 2023, 21(1): e3001983.
[8] WHERRY E J. Molecular Basis of T-Cell Exhaustion[J]. Blood, 2013, 122(21): SCI-38-SCI-38.
[9] CHEN Z, JI Z, NGIOW S F, et al. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision[J]. Immunity, 2019, 51(5): 840-855.e5.
[10] FRANCO F, JACCARD A, ROMERO P, et al. Metabolic and epigenetic regulation of T-cell exhaustion[J]. Nature Metabolism, 2020, 2(10): 1001-1012.
[11] JENKINS E, WHITEHEAD T, FELLERMEYER M, et al. The current state and future of T-cell exhaustion research[J]. Oxford Open Immunology, 2023, 4(1).
[12] CHAN Y T, CHEONG H C, TANG T F, et al. Immune Checkpoint Molecules and Glucose Metabolism in HIV-Induced T Cell Exhaustion[J]. Biomedicines, 2022, 10(11): 2809.
[13] PERALTA R M, XIE B, LONTOS K, et al. Dysfunction of exhausted T cells is enforced by MCT11-mediated lactate metabolism[J]. Nature Immunology, 2024, 25(12): 2297-2307.
[14] PENG M, YIN N, CHHANGAWALA S, et al. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism[J]. Science, 2016, 354(6311): 481-484.
[15] LI F, LIU H, ZHANG D, et al. Metabolic plasticity and regulation of T cell exhaustion[J]. Immunology, 2022, 167(4): 482-494.
[16] RICHTER F C, SALIUTINA M, HEGAZY A N, et al. Take my breath away—mitochondrial dysfunction drives CD8+ T cell exhaustion[J]. Genes & Immunity, 2024, 25(1): 4-6.
[17] VARDHANA S A, HWEE M A, BERISA M, et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen[J]. Nature Immunology, 2020, 21(9): 1022-1033.
[18] PERALTA R, DELGOFFE G. 669 Lactate uptake through MCT11, a novel monocarboxylate transporter, enforces dysfunction in terminally exhausted T cells[C/OL]//Regular and Young Investigator Award Abstracts. BMJ Publishing Group Ltd, 2021: A697-A697.
[19] HUANG Y, SI X, SHAO M, et al. Rewiring mitochondrial metabolism to counteract exhaustion of CAR-T cells[J]. Journal of Hematology & Oncology, 2022, 15(1).
[20] MACIVER N J, MICHALEK R D, RATHMELL J C. Metabolic Regulation of T Lymphocytes[J]. Annual Review of Immunology, 2013, 31(1): 259-283.
[21] YAO C, SUN H W, LACEY N E, et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection[J]. Nature Immunology, 2019, 20(7): 890-901.
[22] DIXON K O, LAHORE G F, KUCHROO V K. Beyond T cell exhaustion: TIM-3 regulation of myeloid cells[J]. Science Immunology, 2024, 9(93).
[23] WANG D, FANG J, WEN S, et al. A comprehensive profile of TCF1+ progenitor and TCF1− terminally exhausted PD-1+CD8+ T cells in head and neck squamous cell carcinoma: implications for prognosis and immunotherapy[J]. International Journal of Oral Science, 2022, 14(1).
[24] XIONG D, ZHANG L, SUN Z J. Targeting the epigenome to reinvigorate T cells for cancer immunotherapy[J]. Military Medical Research, 2023, 10(1).
[25] CHEN Y, XU Z, SUN H, et al. Regulation of CD8+ T memory and exhaustion by the mTOR signals[J]. Cellular & Molecular Immunology, 2023, 20(9): 1023-1039.
[26] CHEN A C Y, JAISWAL S, MARTINEZ D, et al. The aged tumor microenvironment limits T cell control of cancer[J]. Nature Immunology, 2024, 25(6): 1033-1045.
[27] ROSSI M, VECCHI A, TIEZZI C, et al. Phenotypic CD8 T cell profiling in chronic hepatitis B to predict HBV-specific CD8 T cell susceptibility to functional restoration in vitro[J]. Gut, 2023, 72(11): 2123-2137.
[28] GUO M, LIU M Y R, BROOKS D G. Regulation and impact of tumor-specific CD4+ T cells in cancer and immunotherapy[J]. Trends in Immunology, 2024, 45(4): 303-313.
[29] KANG K, LIN X, CHEN P, et al. T cell exhaustion in human cancers[J]. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer, 2024, 1879(5): 189162.
[30] DANIEL B, YOST K E, HSIUNG S, et al. Divergent clonal differentiation trajectories of T cell exhaustion[J]. Nature Immunology, 2022, 23(11): 1614-1627.
[31] Lee J , Lee K , Bae H ,et al.IL-15 promotes self-renewal of progenitor exhausted CD8 T cells during persistent antigenic stimulation[J].Frontiers in Immunology, 2023, 14(14):1117092.
[32] Duque M ,Ana Gírio, Yunes J A ,et al.CASZ1 upregulates PI3K-AKT-mTOR signaling and promotes T-cell acute lymphoblastic leukemia[J].Haematologica, 2023, 109(6):1713-1713.
[33] Huang Y J , Giles J R , Ngiow S F ,et al.1016Enforcement of exhausted T cell epigenetic fate by HMG-box transcription factor TOX[J].Journal for Immunotherapy of Cancer, 2023, 11(Sup1):1.
[34] Rubinstein MP, Kadima AN, Salem ML, Nguyen CL, Gillanders WE, Cole DJ. Systemic administration of IL-15 augments the antigen-specific primary CD8+ T cell response following vaccination with peptide-pulsed dendritic cells[J]. Immunol. 2002 Nov 1;169(9):4928-35.
[35] Ding M, Fei Y, Zhu J, Ma J, Zhu G, Zhen N, Zhu J, Mao S, Sun F, Wang F, Pan Q. IL-27 improves adoptive CD8+ T cells’ antitumor activity via enhancing cell survival and memory T cell differentiation[J]. Cancer Sci. 2022 Jul;113(7):2258-2271.
[36] Salem ML, E El Naggar R, A El Naggar S, A Mobasher M, H Mahmoud M, Badr G. Higher Activities of Hepatic Versus Splenic CD8+ T Cells in Responses to Adoptive T Cell Therapy and Vaccination of B6 Mice with MHC Class-1 Binding Antigen[J]. Iran J Allergy Asthma Immunol. 2017 Dec;16(6):537-553. PMID: 29338160.
[37] Ciraolo E, Althoff S, Ruß J, Rosnev S, Butze M, Pühl M, Frentsch M, Bullinger L, Na IK. Simultaneous Genetic Ablation of PD-1, LAG-3, and TIM-3 in CD8 T Cells Delays Tumor Growth and Improves Survival Outcome[J]. Int J Mol Sci. 2022 Mar 16;23(6):3207.
[38] Hughes DP, Baskar D, Urban FF, Friedman MS, Braun TM, McDonagh KT. Fate and function of anti-CD3/CD28-activated T cells following adoptive transfer: IL-2 promotes development of anti-tumor memory T cells in vivo[J]. Cytotherapy. 2005;7(5):396-407.
[39] Berner VK, duPre SA, Redelman D, Hunter KW. Microparticulate β-glucan vaccine conjugates phagocytized by dendritic cells activate both naïve CD4 and CD8 T cells in vitro[J]. Cell Immunol. 2015 Nov-Dec;298(1-2):104-14.
[40] Alfaro C, Suarez N, Oñate C, Perez-Gracia JL, Martinez-Forero I, Hervas-Stubbs S, Rodriguez I, Perez G, Bolaños E, Palazon A, Sanmamed MF, Morales-Kastresana A, Gonzalez A, Melero I. Dendritic cells take up and present antigens from viable and apoptotic polymorphonuclear leukocytes[J].PLoS One. 2011;6(12):e29300.
[41] Kim JV, Latouche JB, Rivière I, Sadelain M. The ABCs of artificial antigen presentation[J]. Nat Biotechnol. 2004 Apr;22(4):403-10.
[42] Li J, Zhou W, Wang W. Artificial antigen-presenting cells: the booster for the obtaining of functional adoptive cells[J].Cell Mol Life Sci. 2024 Aug 31;81(1):378.

