The immune system, specifically B cells and T cells, plays a complex and dynamic role in myocardial infarction (MI) recovery, influencing both detrimental inflammatory processes and beneficial cardiac repair and remodeling. Following an acute MI, the body initiates a sterile inflammatory response to clear necrotic tissue and promote healing, a process tightly regulated by various immune cell populations.
This review systematically elucidates the immune kinetics following myocardial infarction, focusing on the complex dualistic roles of B and T lymphocytes. It first examines the bifunctional role of B cells in post-infarction healing, where they act as either repair facilitators or inhibitors, and then contrasts the distinct contributions of regulatory and cytotoxic T cells to cardiac repair. Furthermore, it uncovers direct interactions between these lymphocyte subsets within the infarcted myocardium, including the impact of regulatory T cell depletion on B cell function and the mechanisms by which follicular helper T cells drive B cell activation during recovery. Finally, it outlines potential therapeutic strategies aimed at modulating B and T cell responses to enhance post-infarction cardiac repair.
Table of Contents
1. Immune dynamics in myocardial infarction: B and T cell roles
2. B cells in post-MI healing: facilitators or inhibitors?
3. Regulatory versus cytotoxic T cells in cardiac repair
4. Direct interactions between B and T cells in infarcted myocardium
5. Impact of regulatory T cell depletion on B cell function in post-MI inflammation
6. T follicular helper cells drive B cell activation during myocardial infarction recovery
7. Targeting B and T cells to enhance post-infarction repair
01 Immune dynamics in myocardial infarction: B and T cell roles?
The immune dynamics following myocardial infarction (MI) involve a complex interplay of various immune cells, with B and T lymphocytes playing pivotal roles in both the inflammatory response and subsequent cardiac repair or adverse remodeling[1]. Understanding these dynamics is crucial for developing targeted immunomodulatory therapies to improve patient outcomes.
As key mediators of adaptive immunity, T cells exert biphasic functions in the post-myocardial infarction (post-MI) microenvironment: they are primarily pro-inflammatory in the acute phase, with pro-inflammatory Th1 and CD8+ cytotoxic T cells recruited to the infarcted area to release cytokines (e.g., IFN-γ, TNF-α) for necrotic tissue clearance (though prolonged inflammation causes adverse remodeling), while CD4+Foxp3+CD25+ regulatory T cells (Tregs) accumulate in the infarcted myocardium (peaking 3-7 days post-MI) and play a crucial role in immunomodulation and cardiac repair[1,2,3].
B cells, another pivotal component of the adaptive immune system that has historically been less well characterized than T cells in myocardial infarction (MI), display a context‑dependent dual role in post‑MI cardiac recovery, acting either as promoters of adverse remodeling or as mediators of cardioprotective repair. Recent technological advances, especially single‑cell RNA sequencing (scRNA‑seq), have greatly refined our understanding of their functional heterogeneity[4].
On the one hand, B cells contribute to post‑MI pathological progression through multiple mechanisms, including autoantibody‑mediated suppression of cardiac repair, antigen presentation‑driven adverse cardiac remodeling, and alarmin‑induced acceleration of atherosclerosis via plasma cell‑ and immunoglobulin‑dependent pathways. On the other hand, B cells also exert indispensable cardioprotective effects. For example, regulatory B cell subsets facilitate neutrophil apoptosis to attenuate ischemia‑reperfusion injury; in neonatal mice, B cells promote cardiomyocyte proliferation and cardiac regeneration; furthermore, heart‑resident B cells secrete TGF‑β1 to modulate fibroblast activation and scar formation, a process critical for preserving cardiac structural integrity, yet potentially detrimental when dysregulated or excessive[4,5,6].
_.png)
Fig. 1 Immune cells in myocardial infarction (MI)[6].
02 B cells in post-MI healing: facilitators or inhibitors?
As what mentioned before, B cells exhibit a complex, dualistic role in post-myocardial infarction (MI) healing, acting as both facilitators and inhibitors of cardiac repair and remodeling[7].
As facilitators of healing, B cells contribute to myocardial protection. Specific B cell subsets can induce neutrophil apoptosis after ischemia and reperfusion injury, a mechanism that limits tissue damage. Regulatory B cells, a particular subset, are rapidly recruited to the infarcted area and play a protective role. Furthermore, in neonatal mice, B cells have been shown to promote cardiomyocyte proliferation and heart regeneration, suggesting a regenerative capacity in early life[8]. Heart-associated B cells also secrete transforming growth factor-beta 1 (TGF-β1), which participates in fibroblast activation and cardiac fibrosis. While fibrosis is a complex process, scar formation is essential for maintaining cardiac structural integrity post-MI[7,8,9]. B cells are mobilized to the injured myocardium and mediastinal lymph nodes, which drain the heart, via a CXCL13-CXCR5-dependent mechanism, indicating a directed response to injury. They can secrete antibodies and cytokines, thereby regulating immune inflammation and fiber repair processes[10].
Conversely, B cells can also act as inhibitors, contributing to adverse remodeling and exacerbating disease progression. B cell-derived autoantibodies produced in response to MI have been implicated in contributing to ischemic heart failure and hindering post-MI repair. Activated B cells can promote adverse cardiac remodeling in chronic heart failure through antigen presentation. A significant detrimental role is observed where alarmin-activated B cells accelerate atherosclerosis after MI through plasma cell-immunoglobulin-dependent mechanisms. This acceleration of atherosclerosis greatly increases the risk of recurrent cardiovascular events like strokes and further MIs, suggesting a long-term inhibitory effect on cardiovascular health[11].
The activation and functional diversity of B cells post-MI highlight their importance as potential therapeutic targets for immunomodulatory strategies. Understanding the specific subsets of B cells and their cytokine profiles (e.g., IL-10, TGF-β) is critical for developing interventions that can selectively enhance their protective functions while mitigating their detrimental effects. Moreover, changes in blood immune B cells can be utilized to construct risk prediction models for post-MI heart failure, offering diagnostic and prognostic utility. The intricate balance between these pro-healing and pro-disease roles dictates the overall outcome of MI recovery[10,11].
03 Regulatory versus cytotoxic T cells in cardiac repair
In the context of myocardial infarction (MI) recovery, both regulatory T cells (Tregs) and cytotoxic T cells (CD8+ T cells) play distinct, yet interconnected, roles in shaping the inflammatory response, cardiac repair, and subsequent remodeling. While cytotoxic T cells primarily contribute to the initial pro-inflammatory phase and tissue damage, Tregs are crucial for mitigating inflammation and promoting beneficial healing processes[12].
CD8+ cytotoxic T cells are part of the early immune infiltrate following MI. Their role is largely associated with the initial sterile inflammation that characterizes the acute phase of MI. Upon myocardial injury, these cells are recruited to the infarcted area and contribute to the inflammatory cascade. Cytotoxic T cells release pro-inflammatory cytokines, such as interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α), which can exacerbate myocardial injury and potentially contribute to adverse cardiac remodeling. While this initial inflammatory response is necessary for clearing necrotic tissue, an uncontrolled or prolonged cytotoxic T cell response can lead to excessive inflammation and impaired healing. Studies have indicated that the presence of pro-inflammatory T helper 1 (Th1) cells and CD8+ T cells can worsen cardiac function post-MI[12,13].
In contrast to cytotoxic T cells, regulatory T cells (Tregs), characterized by the CD4+Foxp3+CD25+ phenotype, play a pivotal immunomodulatory and cardioprotective role during recovery from myocardial infarction (MI). Following MI, Tregs differentiate and accumulate rapidly within the infarcted myocardium, a process actively supported by the local cardiac microenvironment. Their numbers typically peak between 3 and 7 days post-MI in both the ischemic myocardium and mediastinal lymph nodes[14].
The cardioprotective functions of Tregs in cardiac repair and remodeling are multifaceted. First, they suppress excessive infiltration of inflammatory cells and dampen immune responses through the secretion of anti-inflammatory cytokines, primarily interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), which can be quantified using IL-10 assays and TGF-β assays in experimental studies. This cytokine-mediated regulation mitigates prolonged inflammation and prevents adverse left ventricular remodeling. Second, Tregs reduce oxidative stress within the infarcted tissue, which helps protect cardiomyocytes from acute injury. Third, they modulate fibrotic processes: while basal fibrosis is necessary for scar formation and structural integrity, Tregs limit pathological excessive fibrosis that could otherwise lead to myocardial stiffening and dysfunction. Fourth, Tregs promote angiogenesis, facilitating the formation of new blood vessels essential for restoring perfusion and supporting tissue regeneration in ischemic areas. In addition, Tregs interact with other immune cell populations, such as macrophages, to drive a phenotypic switch toward a pro-reparative state, ultimately resolving inflammation and enhancing tissue repair. Notably, in neonatal hearts, which possess a high intrinsic regenerative capacity, Tregs also contribute to fibrosis regulation and cardiac repair, thereby underscoring their broader relevance in myocardial regeneration[13,14].
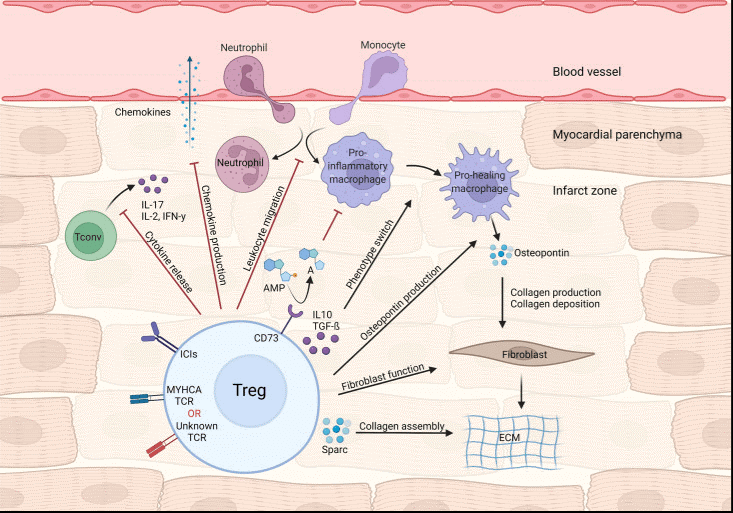
Fig. 2 Treg-mediated effects in myocardial repair and inflammation after infarction[15].
%20cells%20in%20Peripheral%20blood%20mononuclear%20cells%20(PBMCs)_.png)
Fig. 3 Detection of regulatory T (TREG) cells in Peripheral blood mononuclear cells (PBMCs). PBMCs were stained with Elab Fluor® Violet 450 anti human CD45, Elab Fluor® Red 780 anti human CD3, FITC anti human CD4, PerCP/Cyanine5.5 anti human CD8, PE anti human CD25, and APC anti human CD127, followed by analysis via flow cytometry. Regulatory T (Treg) cells display the phenotype CD45+CD3+CD4+CD127low/-CD25+. (The data are provided by Elabscience.)
%20cells%20in%20in%20splenocytes%20from%20C57BL%206%20mice_.png)
Fig. 4 Detection of regulatory T (TREG) cells in in splenocytes from C57BL/6 mice. Splenocytes were surface stained with Elab Fluor® Violet 450 Anti-Mouse CD45, PE/Cyanine7 Anti-Mouse CD45R/B220, FITC Anti-Mouse CD3, Elab Fluor® Red 780 Anti-Mouse CD4, PerCP Anti-Mouse CD8a and APC Anti-Mouse CD25 and then treated with Foxp3/Transcription Factor Staining Kit. Cells were then stained with PE Anti-Mouse Foxp3, followed by analysis via flow cytometry. Regulatory T cells(Treg cells) display the phenotype CD45+CD45R/B220-CD3+CD4+CD25+Foxp3+. (The datas are provided by Elabceience.)
Elabscience® Quick Overview of Popular Products:
Table 1. Multicolor Panel for Flow Cytometric Analysis of Human and Mouse Regulatory T cells (Tregs)
|
Marker |
Clone |
Fluorochrome |
Cat. No. |
Species Reactivity |
|
CD45 |
HI30 |
Elab Fluor® Violet 450 |
E-AB-F1137Q |
Human |
|
CD3 |
UCHT1 |
Elab Fluor® Red 780 |
E-AB-F1230S |
Human |
|
CD4 |
SK3 |
FITC |
E-AB-F1352C |
Human |
|
CD8 |
OKT-8 |
PerCP/Cyanine5.5 |
E-AB-F1110J |
Human |
|
CD25 |
BC96 |
PE |
E-AB-F1194D |
Human |
|
CD127 |
A019D5 |
APC |
E-AB-F1152E |
Human |
|
Foxp3 |
FJK-16s |
PE |
E-AB-F1351D |
Mouse |
|
CD45R/B220 |
RA3.3A 1/6.1 |
PE/Cyanine7 |
E-AB-F1112H |
Mouse |
|
CD25 |
PC-61.5.3 |
APC |
E-AB-F1102E |
Mouse |
|
CD4 |
GK1.5 |
Elab Fluor® Red 780 |
E-AB-F1097S |
Mouse |
|
CD45 |
30-F11 |
Elab Fluor® Violet 450 |
E-AB-F1136Q |
Mouse |
|
CD3 |
17A2 |
FITC |
E-AB-F1013C |
Mouse |
|
CD8a |
53-6.7 |
PerCP |
E-AB-F1104F |
Mouse |
04 Direct interactions between B and T cells in infarcted myocardium
In the context of myocardial infarction (MI), the direct interactions between B and T cells within the infarcted myocardium are crucial for shaping the immune response and influencing subsequent cardiac repair and remodeling. While both cell types have distinct primary functions, their cross-talk significantly modulates the local microenvironment.
The direct interactions between B and T cells within the infarcted myocardium involve several mechanisms. Antigen-presenting cells (APCs), which include B cells, present cardiac antigens to T cells, influencing their activation and differentiation. B cells can interact with T cells, leading to cytokine production that either exacerbates or resolves inflammation. For instance, B cells can produce cytokines like IL-10 that influence Treg function, or pro-inflammatory cytokines that enhance T cell-mediated inflammation. Conversely, T cells can regulate B cell activation and antibody production. This interplay is critical for maintaining the balance between adaptive immunity and self-tolerance in the damaged heart. The local myocardial environment, including specific cytokines and chemokines, influences the recruitment and function of both B and T cells, further modulating their interactions. The lymphatic system also plays a crucial role in regulating post-MI inflammation by interacting with immune cells, influencing their clearance and surveillance, which indirectly affects B and T cell dynamics within the heart[15].
Understanding these direct interactions is pivotal for developing immunomodulatory therapies. Strategies aimed at tipping the balance towards pro-healing B and T cell phenotypes, such as enhancing Treg function or selectively inhibiting detrimental B cell subsets, hold promise for improving MI recovery outcomes. The use of advanced techniques like single-cell RNA sequencing is providing detailed insights into these cellular dynamics, revealing persistent transcriptional changes in cardiac adaptive immune cells post-MI and identifying potential targets for intervention[16].
_.png)
Fig. 5 Detection of TBNK cell subsets in human peripheral blood mononuclear cells (PBMCs). PBMCs were stained with Anti-HumanCD19-FITC/CD56-PE/CD3-PE/Cyanine7/CD45-PerCP Cocktail (E-AB-FC0011), and subsequently analyzed by flow cytometry. (The data are provided by Elabscience.)
Elabscience® Quick Overview of Popular Products:
Table 2. Reagents for T cells and B cells Research
|
Product Name |
Cat. No. |
|
EasySort™ Mouse CD8+T Cell Isolation Kit |
MIM003N |
|
EasySort™ Mouse CD3+T Cell Isolation Kit |
MIM001N |
|
EasySort™ Mouse CD4+T Cell Isolation Kit |
MIM002N |
|
EasySort™ Human CD3+T Cell Isolation Kit |
MIH001N |
|
EasyStain™ Human Fc Receptor Blocking Solution |
E-CK-A171 |
|
STYX™ Violet Fixable Viability Kit |
E-CK-A167 |
|
STYX™ Near-IR Fixable Viability Kit |
E-CK-A168 |
|
STYX™ Green Fixable Viability Kit |
E-CK-A166 |
|
STYX™ Red Fixable Viability Kit |
E-CK-A170 |
|
STYX™ Yellow Fixable Viability Kit |
E-CK-A169 |
|
Anti-Human CD45-FITC/CD3-PE/Cyanine5/CD56/NCAM-PE/CD19-PE/Elab Fluor® 594 Cocktail |
E-AB-FC0028 |
|
Anti-Human CD45-FITC/CD3-PE/Cyanine5/CD4-PE/CD8-PE/Elab Fluor® 594 Cocktail |
E-AB-FC0029 |
|
Anti-Human CD3-FITC/CD19-PE Cocktail |
E-AB-FC0004 |
|
Anti-Human CD3-FITC/CD16+CD56-PE Cocktail |
E-AB-FC0001 |
|
Anti-Human CD3-APC/CD4-FITC/CD8a-PE Cocktail |
E-AB-FC0008 |
|
Anti-Human CD3-FITC/CD4-PE/CD45-PerCP-Cyanine5.5 Cocktail |
E-AB-FC0003 |
|
Anti-Human CD19-FITC/CD56-PE/CD3-PE/Cyanine7/CD45-PerCP Cocktail |
E-AB-FC0011 |
05 Impact of regulatory T cell depletion on B cell function in post-MI inflammation
Depletion of regulatory T cells (Tregs) exerts a significant impact on B cell function following myocardial infarction (MI). Specifically, this Treg depletion is primarily characterized by enhanced activation of pro-inflammatory B cell subsets, impaired functional activity of regulatory B cells (Bregs), and deterioration of the myocardial inflammatory microenvironment, which collectively lead to aggravated myocardial injury and accelerated adverse cardiac remodeling[16].
5.1 Impact of Treg Depletion on B Cell Activation
Regulatory T cells (Tregs) play a crucial role in maintaining immune homeostasis following myocardial infarction (MI) by suppressing inflammatory cell infiltration and modulating macrophage polarization[16]. Upon Treg depletion, the pro-inflammatory microenvironment becomes dysregulated, which in turn triggers the activation of B cells that secrete CCL7[17]. This chemokine robustly recruits Ly6C+ monocytes to the infarcted myocardium, thereby exacerbating the local inflammatory response. Accumulating evidence has demonstrated that B cells accumulate in the heart during the acute phase of MI and contribute to pathological myocardial fibrosis through the production of pro-inflammatory cytokines. Specifically, Treg depletion removes the regulatory constraints on this process, enabling uncontrolled B cell-driven pro-inflammatory activity that further expands infarct size and aggravates cardiac dysfunction[17].
5.2 Reciprocal Regulatory Relationship Between Tregs and Bregs
Tregs and regulatory B cells (Bregs) collaboratively maintain immune tolerance in the post-MI heart. Specifically, Bregs promote the activation of Tregs, which in turn sustain long-term immunomodulatory effects. Depletion of Tregs disrupts this delicate equilibrium, thereby resulting in impaired Breg function. Clinical studies have shown that the frequency of circulating CD24hiCD38hi Bregs is significantly lower in patients with MI compared to those with stable angina. Furthermore, the loss of Tregs further suppresses the generation and maintenance of Bregs, reducing their capacity to inhibit the activation of conventional T cells and macrophages through both IL-10 secretion and contact-dependent mechanisms. Although CD5+ Bregs enriched in pericardial adipose tissue exert anti-inflammatory effects to attenuate post-MI inflammation, Treg depletion undermines this local regulatory mechanism[18].
5.3 Mechanisms Underlying Worsening of the Inflammatory Microenvironment
Tregs sustain an anti-inflammatory milieu in the post-MI heart through multiple pathways, including inhibiting neutrophil and monocyte infiltration, inducing anti-inflammatory macrophage polarization, and promoting M2 macrophage differentiation via exosome secretion. Following Treg depletion, a profound shift toward pro-inflammatory macrophage dominance occurs. Concurrently, B cells, released from Treg-mediated suppression, augment their pro-inflammatory functions, including the secretion of TNF-α, IL-1β, and IL-6. In addition, Treg deficiency disrupts the Th1/Th2 balance and elevates the Th17/Treg ratio, which synergizes with B cell-mediated pro-inflammatory responses to further exacerbate myocardial inflammation. Specifically, studies have confirmed that mice subjected to Treg depletion 8 weeks post-MI exhibit an increased Th17/Treg ratio and worsened cardiac remodeling[19].
5.4 Acceleration of Myocardial Fibrosis
Although Tregs themselves can promote fibrosis by enhancing collagen deposition, their depletion leads to uncontrolled inflammation and excessive B cell activation, which more severely aggravates fibrotic remodeling. Previous studies have shown that B cell depletion via CD20 blockade inhibits pressure overload-induced cardiac remodeling. In contrast, Treg depletion enhances B cell activation and thereby promotes TGF-β-induced myocardial fibrosis. Moreover, the absence of Tregs diminishes the protection against cardiomyocyte apoptosis, while B cell-driven pro-inflammatory responses further exacerbate cardiomyocyte death, collectively accelerating the progression of pathological myocardial fibrosis[20].
06 T follicular helper cells drive B cell activation during myocardial infarction recovery
Direct evidence implicating follicular helper T (Tfh) cells in driving B cell activation during the recovery phase of myocardial infarction (MI) remains limited. To date, our understanding of this process is largely derived from correlative clinical observations, where elevated levels of circulating Tfh (cTfh) cells in peripheral blood correlate with the severity of acute MI (AMI) and adverse clinical outcomes. However, studies specifically investigating Tfh-mediated mechanisms during the post-MI recovery period are relatively scarce. Existing literature has demonstrated that CD4+ T cell subsets, particularly T helper 1 (Th1) cells and regulatory T cells (Tregs), are the predominant mediators of myocardial repair following MI, whereas accumulating B cells confer protective effects via the production of interleukin-10 (IL-10). In contrast, Tfh cells are more frequently reported in the context of chronic transplant rejection, where they facilitate the differentiation of B cells into plasma cells[21].
6.1 Immune Dynamics During Post-MI Recovery and the Tfh-B Cell Axis
The post-MI immune response proceeds through three sequential and overlapping phases: the acute inflammatory phase, characterized by the infiltration of neutrophils and pro-inflammatory M1 macrophages; the reparative phase, dominated by M2 macrophages and Tregs; and the scar formation phase. Within 45 minutes of myocardial reperfusion, CD4+ T cells infiltrate the myocardium in response to cardiac self-antigens (e.g., myosin heavy chain alpha [MYHCA]), thereby promoting monocyte recruitment and differentiation and contributing to favorable cardiac remodeling. In Rag1⁻/⁻ mice subjected to myocardial ischemia/reperfusion (MI/R) injury, reduced myocardial inflammation and smaller infarct sizes are observed; however, reconstitution with interferon-γ (IFN-γ)-producing CD4+ T (Th1) cells reverses this protective phenotype, underscoring the injurious role of Th1 cells in post-MI pathology. As a distinct CD4⁺ T cell subset, Tfh cells provide critical signals, including CD40 ligand (CD40L), programmed cell death protein 1 (PD-1), and IL-21, that support B cell proliferation, affinity maturation, and immunoglobulin class switching. Nevertheless, the specific role of Tfh cells in germinal center (GC) formation during the MI recovery phase has not been clearly defined in the existing MI-related literature[22].
Increased frequencies of circulating T follicular helper (cTfh) cells have been consistently observed in patients with acute myocardial infarction (AMI). Elevated cTfh cell levels positively correlate with the proportions of CD3⁺CD8⁺ effector memory T cells and are significantly associated with disease severity and in-hospital adverse outcomes (rho = 0.155, P = 0.002)[23]. Tfh-associated factors, including C-X-C motif chemokine ligand 13 (CXCL13) and IL-21, facilitate the differentiation of B cells into plasmablasts and class-switched memory B cells, thereby enhancing immunoglobulin G (IgG) production. Notably, these findings differ from those observed in the chronic phase of heart transplantation, where the expansion of cTfh17 subsets and the enhanced anti-apoptotic properties of cTfh2 subsets drive antibody-mediated rejection (AMR) and cardiac allograft vasculopathy, a pathophysiological context distinct from that of acute MI. Following MI, accumulating B cells preferentially secrete IL-10, which attenuates myocardial inflammation and preserves cardiac function, indicating a predominantly protective role of B cells in the post-MI setting[24].
6.2 Inferred Mechanisms of Tfh Cell Involvement
Myocardial injury releases cardiac self-antigens that activate heart-specific CD4+ T cells. Adoptive transfer of MYHCA-specific T cell receptor (TCR)-M cells leads to their accumulation in the infarcted myocardium and mediastinal lymph nodes, where these cells acquire a Treg phenotype and promote tissue healing. Tfh cell differentiation depends on B cell-mediated antigen presentation and inducible T cell co-stimulator ligand (ICOS-L) co-stimulation, which provide compensatory signals supporting follicular migration and GC reactions. During the post-MI recovery phase, persistent exposure to cardiac self-antigens may induce cTfh cell expansion, which is analogous to the mechanisms observed in chronic transplant rejection, thereby potentially activating B cells within lymphoid organs via IL-21 and CXCL13 to generate memory B cells and plasma cells. However, in the post-MI setting, Tregs and Foxp3+CD4+ T cells predominantly drive the M1-to-M2 macrophage transition, a process that appears to override Tfh-mediated humoral immunity. Furthermore, stem cell-based therapeutic approaches have been shown to modulate T cell proliferation without altering B and T cell infiltration at 7 days post-MI, suggesting that interventions targeting adaptive immunity may exert differential temporal and subset-specific effects on post-MI immune responses[22,23,24].
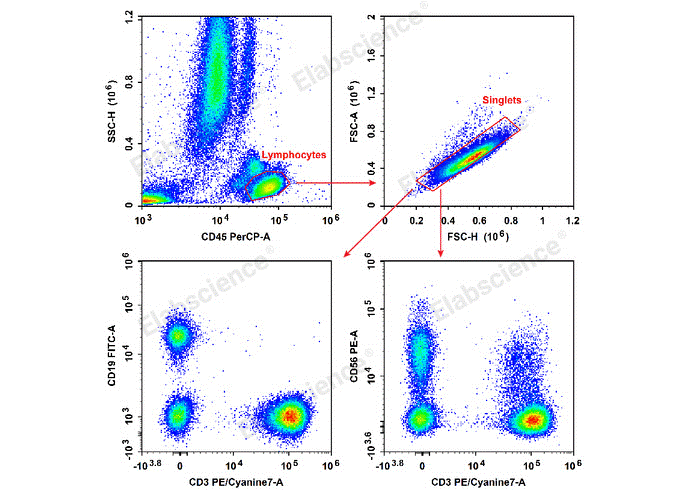
Fig. 5 Detection of Th1 and Th2 cell subsets in human peripheral blood mononuclear cells (PBMCs). PBMCs were stimulated with Cell Stimulation Mix and Protein Transport Inhibitor Mix for 5 hours, then harvested, fixed, and permeabilized. Subsequently, cells were stained with PerCP/Cyanine5.5 Anti-Human CD3, Elab Fluor® 488 Anti-Human CD4, PE Anti-Human IL-4 and APC Anti-Human IFN-γ to assess the proportions of Th1 and Th2 cells, as well as their capacity to produce the effector cytokines IFN-γ and IL-4, respectively. Th1 cells were identified as CD3+CD4+IFN-γ+, and Th2 cells as CD3+CD4+IL-4+. (The data are provided by Elabscience.)
Elabscience® Quick Overview of Popular Products:
Table 3. Multicolor Panel for Flow Cytometric Analysis of Human and Mouse Helper T cells (CD4+ T cells)
|
Marker |
Clone |
Fluorochrome |
Cat. No. |
Species Reactivity |
|
CD3 |
OKT-3 |
PerCP/Cyanine5.5 |
E-AB-F1001J |
Human |
|
CD4 |
SK3 |
Elab Fluor® 488 |
E-AB-F1352L |
Human |
|
IL-17A |
BL168 |
PE |
E-AB-F1173D |
Human |
|
IFN-γ |
B27 |
APC |
E-AB-F1196E |
Human |
|
IL-4 |
MP4-25D2 |
PE |
E-AB-F1203D |
Human |
|
CD3 |
17A2 |
PE/Cyanine5 |
E-AB-F1013G |
Mouse |
|
CD4 |
GK1.5 |
FITC |
E-AB-F1097C |
Mouse |
|
IFN-γ |
XMG1.2 |
APC |
E-AB-F1101E |
Mouse |
|
IL-4 |
11B11 |
PE |
E-AB-F1204D |
Mouse |
|
IL-17A |
17F3 |
PE |
E-AB-F1272D |
Mouse |
07 Targeting B and T cells to enhance post-infarction repair
Therapeutic strategies targeting B cells and T cells following myocardial infarction (MI) necessitate precise modulation of their dualistic roles, as these adaptive immune cells can either promote repair or exacerbate injury. Current evidence indicates that expanding regulatory T cells (Tregs) and activating protective B cell subsets represent key approaches for enhancing myocardial repair, while suppressing pro-inflammatory T cells (Th1) and pro-fibrotic B cell responses may limit adverse remodeling.
7.1 Therapeutic Strategies Targeting Treg Expansion
Low-dose interleukin-2 (IL-2) therapy is currently the most promising Treg-targeted strategy in clinical trials. By selectively expanding and activating Tregs while suppressing effector T cells, this approach is anticipated to promote post-MI cardiac repair and limit atherosclerosis progression. The rationale for this strategy stems from the discovery of heart-specific Tregs: in murine MI models, CD4+ T cells recognizing cardiac myosin heavy chain alpha (MYHCA) migrate to the infarcted myocardium, acquire a Treg phenotype, and exhibit a distinct pro-repair gene expression profile that mediates cardioprotection.
7.2 Strategies for Modulating B Cell Activation
Bruton tyrosine kinase (BTK) inhibitors, such as ibrutinib, improve post-MI cardiac function by suppressing bone marrow B cell development and peripheral lymphoid organ B cell activation. Specifically, this strategy targets the pro-fibrotic and pro-inflammatory effects of B cells, particularly during the chronic phase of cardiac remodeling. Given that B cell functions are model-dependent, distinct cardiac B cell subsets exhibit diverse roles across experimental models. Therefore, therapeutic approaches should distinguish between protective (IL-10-producing) and detrimental (pro-fibrotic cytokine-producing) B cells, prioritizing preservation or expansion of the former[28].
7.3 Translational Strategies for Time-Dependent Immunomodulation
Studies have revealed opposing roles of CD4+ T cells across different MI phases. Specifically, during the acute phase (0-7 days post-MI), CD4+ T cells promote myocardial repair, whereas during the chronic phase (4-8 weeks post-MI), they contribute to adverse cardiac remodeling and excessive fibrosis. Consequently, temporal immunomodulation is a feasible translational strategy, involving promotion of Tregs and protective B cells in the early phase and suppression of Th1 cells and pro-fibrotic B cells in the late phase[29].
In addition to temporal immunomodulation, chimeric antigen receptor (CAR)-T cell therapy targeting fibroblast activation protein (FAP) reduces myocardial fibrosis and improves both systolic and diastolic cardiac function. Similarly, bone marrow mesenchymal stem cells (BM-MSCs) promote myocardial repair by inhibiting T cell proliferation, modulating adaptive immune responses, and facilitating monocyte and macrophage recruitment; these two approaches represent additional translationally promising strategies for post-MI treatment[30].
In conclusion, cardiac-draining lymph nodes (med-LNs) are enlarged in MI patients with increased cellularity, and these changes correlate with infarct size and cardiac function, suggesting lymphoid organs as potential immunomodulatory targets. However, excessive suppression of inflammatory responses is unfeasible, as acute-phase immune cell infiltration is essential for proper infarct healing and ventricular structure-function preservation. Thus, future research should further elucidate direct interaction mechanisms between T cells and cardiac fibroblasts, subset-specific B cell functions, and optimal immunomodulatory therapeutic time windows to maximize efficacy while avoiding adverse effects[31,32].
References:
[1] Delgobo M, Weiß E, Ashour D E D, et al. Myocardial milieu favors local differentiation of regulatory T cells[J]. Circulation research, 2023, 132(5): 565-582.
[2] Huang F, Zhang J, Zhou H, et al. B cell subsets contribute to myocardial protection by inducing neutrophil apoptosis after ischemia and reperfusion[J]. JCI insight, 2024, 9(4): e167201.
[3] Yang Z, Zhang Z, Feng S, et al. Cross-talk between cardiac lymphatics and immune cells regulates inflammatory response and cardiac recovery after myocardial infarction[J]. Frontiers in immunology, 2025, 16: 1557250.
[4] Heinrichs M, Ashour D E D, Siegel J, et al. The healing myocardium mobilizes a distinct B-cell subset through a CXCL13-CXCR5-dependent mechanism[J]. Cardiovascular Research, 2021, 117(13): 2664-2676.
[5] Kyaw T, Loveland P, Kanellakis P, et al. Alarmin-activated B cells accelerate murine atherosclerosis after myocardial infarction via plasma cell-immunoglobulin-dependent mechanisms[J]. European heart journal, 2021, 42(9): 938-947.
[6] Feng Q, Li Q, Zhou H, et al. The role of major immune cells in myocardial infarction[J]. Frontiers in Immunology, 2023, 13: 1084460.
[7] Jin K, Gao S, Yang P, et al. Single‐cell RNA sequencing reveals the temporal diversity and dynamics of cardiac immunity after myocardial infarction[J]. Small Methods, 2022, 6(3): 2100752.
[8] Huang F, Zhang J, Zhou H, et al. B cell subsets contribute to myocardial protection by inducing neutrophil apoptosis after ischemia and reperfusion[J]. JCI insight, 2024, 9(4): e167201.
[9] Ma J, Wang X, Jia Y, et al. The roles of B cells in cardiovascular diseases[J]. Molecular Immunology, 2024, 171: 36-46.
[10] Satarkar D, Patra C. Evolution, expression and functional analysis of CXCR3 in neuronal and cardiovascular diseases: a narrative review[J]. Frontiers in Cell and Developmental Biology, 2022, 10: 882017.
[11] Tan Y, Duan X, Wang B, et al. Murine neonatal cardiac B cells promote cardiomyocyte proliferation and heart regeneration[J]. npj Regenerative Medicine, 2023, 8(1): 7.
[12] de Winter N, Ji J, Sintou A, et al. Persistent transcriptional changes in cardiac adaptive immune cells following myocardial infarction: New evidence from the re-analysis of publicly available single cell and nuclei RNA-sequencing data sets[J]. Journal of molecular and cellular cardiology, 2024, 192: 48-64.
[13] Kologrivova I, Shtatolkina M, Suslova T, et al. Cells of the immune system in cardiac remodeling: main players in resolution of inflammation and repair after myocardial infarction[J]. Frontiers in immunology, 2021, 12: 664457.
[14] Zhang Y, Wen W, Liu H. The role of immune cells in cardiac remodeling after myocardial infarction[J]. Journal of cardiovascular pharmacology, 2020, 76(4): 407-413.
[15] Farache Trajano L, Smart N. Immunomodulation for optimal cardiac regeneration: insights from comparative analyses[J]. NPJ Regenerative medicine, 2021, 6(1): 8.
[16] Qin Y, Li M, Liu H. Regulatory T cells: a promising new therapeutic target in ventricular remodeling after myocardial infarction[J]. Frontiers in Immunology, 2025, 16: 1514335.
[17] Lu Y, Xia N, Cheng X. Regulatory T cells in chronic heart failure[J]. Frontiers in immunology, 2021, 12: 732794.
[18] Jansen K, Cevhertas L, Ma S, et al. Regulatory b cells, a to z[J]. Allergy, 2021, 76(9): 2699-2715.
[19] Alshoubaki Y K, Nayer B, Lu Y Z, et al. Tregs delivered post-myocardial infarction adopt an injury-specific phenotype promoting cardiac repair via macrophages in mice[J]. Nature communications, 2024, 15(1): 6480.
[20] Zou A E, Kongthong S, Mueller A A, et al. Fibroblasts in immune responses, inflammatory diseases and therapeutic implications[J]. Nature Reviews Rheumatology, 2025, 21(6): 336-354.
[21] Bi X, Zhang Z, Zhang Z, et al. Peripheral blood memory T cells and follicular helper T cells as potential biomarkers for assessing disease severity and in-hospital outcomes in acute myocardial infarction patients[J]. American Journal of Translational Research, 2025, 17(9): 7120.
[22] Hofmann U, Frantz S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction[J]. Circulation research, 2015, 116(2): 354-367.
[23] Bi X, Zhang Z, Zhang Z, et al. Peripheral blood memory T cells and follicular helper T cells as potential biomarkers for assessing disease severity and in-hospital outcomes in acute myocardial infarction patients[J]. American Journal of Translational Research, 2025, 17(9): 7120.
[24] Zeng X, Pan Y, Xia Q, et al. The effects of interleukin-21 in the biology of transplant rejection[J]. Frontiers in Immunology, 2025, 16: 1571828.
[25] Hofmann U, Frantz S. Role of T-cells in myocardial infarction[J]. European heart journal, 2016, 37(11): 873-879.
[26] Wang Y, Wang C, Shen L, et al. The role of regulatory T cells in heart repair after myocardial infarction[J]. Journal of cardiovascular translational research, 2023, 16(3): 590-597.
[27] Wang Y, et al. "Regulatory T lymphocytes in myocardial infarction: a promising new therapeutic target." International Journal of Cardiology 203 (2016): 923-928.
[28] von Hundelshausen P, Siess W, Duan R, et al. Involvement of Btk in Cardiovascular Disease and Its Therapeutic Targeting[J]. Circulation, 2026, 153(6): 435-456.
[29] Yuan D, Tie J, Xu Z, et al. Dynamic Profile of CD4+ T‐Cell‐Associated Cytokines/Chemokines following Murine Myocardial Infarction/Reperfusion[J]. Mediators of Inflammation, 2019, 2019(1): 9483647.
[30] Meng S, Hara T, Miura Y, et al. Fibroblast activation protein constitutes a novel target of chimeric antigen receptor T‐cell therapy in solid tumors[J]. Cancer Science, 2024, 115(11): 3532-3542.
[31] Brakenhielm E, Alitalo K. Cardiac lymphatics in health and disease[J]. Nature Reviews Cardiology, 2019, 16(1): 56-68.
[32] Gottumukkala R V, Lv H J, Cornivelli L, et al. Myocardial infarction triggers chronic cardiac autoimmunity in type 1 diabetes[J]. Science translational medicine, 2012, 4(138): 138ra80-138ra80.

