Dyslipidemia is a key driver of myocardial infarction (MI) through its role in coronary artery disease (CAD). Atherosclerosis, the underlying pathology, initiates when LDL promotes endothelial dysfunction and vascular inflammation, leading to plaque formation within coronary arteries. This progressive narrowing and instability characterize CAD. Insulin resistance exacerbates this cascade, inducing a pro-atherogenic profile that accelerates the formation of rupture-prone plaques. Plaque rupture triggers thrombosis, resulting in acute MI.
In this context, we highlight the central role of lipid metabolism as a critical and modifiable axis in the pathophysiology of CAD and MI. Correcting this dysfunction through aggressive LDL-lowering strategies and targeted interventions for insulin resistance represents a proven therapeutic approach to stabilize atherosclerotic plaques, mitigate disease progression, and significantly reduce the risk of adverse cardiovascular events.
Table of Contents
1. Dysregulated lipid metabolism as a driver of myocardial infarction risk
2. Mechanisms linking lipid metabolism disorders to myocardial infarction
3. LDL cholesterol in lipid dysregulation and myocardial infarction pathophysiology
4. Biomarkers of lipid dysfunction predictive of myocardial infarction
5. Insulin resistance modulates lipid metabolism and cardiovascular risk
6. Elevated free fatty acids promote cardiac ischemia and infarction in metabolic disorders
01 Dysregulated lipid metabolism as a driver of myocardial infarction risk
Abnormal lipid metabolism is a fundamental, causal and modifiable risk factor for MI[1].Epidemiological and Mendelian randomization studies have verified that genetic predisposition to atherogenic dyslipidemia directly elevates MI risk independent of environmental confounders. At the molecular level, genes like Plasminogen Activator Urokinase Receptor (PLAUR) and QKI (differentially expressed in acute MI) participate in pathways such as lipophagy; their disruption leads to macrophage lipid accumulation, foam cell formation, and accelerated atherosclerosis[2].
Therefore, abnormal lipid metabolism is not merely correlated but is a primary pathogenic driver, positioning it decisively among the most critical myocardial infarction risk factors.
02 Mechanisms linking lipid metabolism disorders to myocardial infarction
Lipid metabolism disorders initiate atherosclerosis via subendothelial retention and modification of LDL[3-5]. Clinical LDL assays are essential for accurately quantifying LDL levels, which helps identify individuals at higher risk of plaque formation. Modified LDL induces endothelial dysfunction and monocyte recruitment, followed by macrophage differentiation.These macrophages then internalize modified LDL to form foam cells and generate fatty streaks. Sustained inflammation and atherogenic dyslipidemia drive plaque progression, forming a lipid-rich necrotic core and a fibrous cap[6]. Lipid disorders exacerbate plaque vulnerability by enlarging the necrotic core and promoting inflammatory secretion of proteolytic enzymes that degrade the cap, making it prone to rupture[7]. Plaque rupture or erosion exposes thrombogenic material, triggering occlusive thrombus formation and resulting in myocardial infarction.Thus, these disorders are central to initiating atherosclerosis, sustaining inflammation, and precipitating acute thrombosis.
03 LDL cholesterol in lipid dysregulation and myocardial infarction pathophysiology
Low-density lipoprotein cholesterol (LDL-C) is the primary atherogenic agent in dyslipidemia, with plasma levels showing a linear, causal relationship with MI risk. Pathogenicity depends on both particle quantity and quality, particularly oxidative modification within the arterial intima. Following subendothelial retention, LDL undergoes oxidation and is internalized by macrophages via scavenger receptors such as LOX-1 and CD36[8]. This uptake drives foam cell formation and triggers a pro-inflammatory response that accelerates plaque progression. Highly oxidation-prone small, dense LDL (sdLDL) particles further amplify this process. Consequently, serum LDL-C, a key component of the serum cholesterol range, remains the principal biomarker and therapeutic target for MI prevention[9]. Accurate measurement of LDL-C through clinical LDL assays and routine LDL tests is essential for monitoring cardiovascular risk. Substantial clinical evidence confirms that lowering LDL-C, especially in high-risk populations such as patients with type 2 diabetes, proportionally decreases cardiovascular events[10].
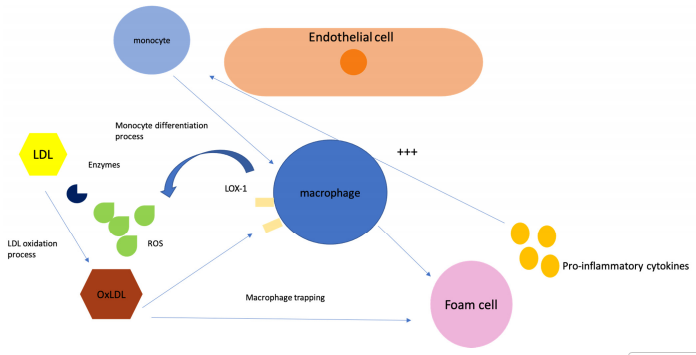
Fig. 1 Oxidation of LDL-C in the sub-endothelium[8]. Macrophages take up oxLDL via scavenger receptors (LOX-1, CD36), leading to foam cell formation and inflammation, which drives plaque progression.
04 Biomarkers of lipid dysfunction predictive of myocardial infarction
Beyond conventional lipid panels (e.g., total cholesterol, LDL-C, HDL-C), advanced multi-omics approaches have identified novel biomarkers for MI risk associated with lipid metabolism dysfunction. Lipidomic analyses have identified specific phosphatidylethanolamine species (e.g., PE(37:4), PE(17:0/20:4)) that gradually increase from healthy individuals to acute MI patients and act as independent predictors[11, 12]. Persistent changes in ceramide and sphingomyelin levels after MI are associated with adverse cardiac remodeling. Moreover, elevated lipoprotein(a) and oxidized phospholipids (OxPL-apoB, OxPL-apo(a)) can predict the severity of coronary artery disease, while genes such as TLR2 and S100A9 are recognized as potential diagnostic biomarkers[13]. Chronic elevation of plasma free fatty acids is a prognostic biomarker for sudden cardiac death post-MI[14]. Advanced carotid atherosclerosis in metabolic disorders reflects the interplay of inflammation, insulin resistance, and dyslipidemia, highlighting multifactorial biomarker profiles[15].
05 Insulin resistance modulates lipid metabolism and cardiovascular risk
Insulin resistance (IR) profoundly disturbs lipid metabolism, creating a pro-atherogenic milieu that increases myocardial infarction (MI) incidence[16]. Hepatic insulin resistance, as measured by clinical insulin assays, enhances de novo lipogenesis and VLDL secretion, leading to hypertriglyceridemia[16]. This promotes cholesteryl estertransfer protein (CETP)-mediated lipid exchange, resulting in the generation of small, dense LDL and a reduction in HDL-C levels[17].
Concurrently, insulin resistance in adipose tissue elevates plasma free fatty acids, further driving VLDL production and impairing endothelial function. The bidirectional relationship between inflammation and insulin resistance involves pro-inflammatory cytokines disrupting insulin signaling (e.g., PI3K/Akt), while hyperinsulinemia and lipotoxicity activate innate immune pathways, fueling plaque instability[18]. This cluster defines insulin resistance and metabolic syndrome, elevating MI risk by approximately 1.7 fold[19].
In type 2 diabetes, insulin resistance leads to a characteristic lipoprotein phenotype with elevated apolipoprotein B-containing lipoproteins, hypertriglyceridemia, low HDL-C, and increased small, dense LDL[20]. Agents such as glucagon-like peptide 1(GLP-1) receptor agonists ameliorate GLP-1 insulin resistance pathways, reducing MI risk through multifaceted cardiometabolic benefits[21].
_.png)
Fig. 2 Schematic depiction of the pathophysiological factors linking IR with cardiovascular disease (CVD)[16]. Elevated FFA levels lead to lipotoxicity, inflammation, and dyslipidemia. Hyperglycemia and IR promote oxidative stress, resulting in the production of reactive oxygen species (ROS), oxLDL-C, and advanced glycation end-products (AGEs), which in turn reduce glucose uptake by tissues. These processes collectively contribute to endothelial dysfunction and hypertension, ultimately leading to the development of CVD.
06 Elevated free fatty acids promote cardiac ischemia and infarction in metabolic disorders
In patients with metabolic disorders, elevated plasma free fatty acids worsen cardiac ischemia and affect MI prognosis via multiple mechanisms. Sustained high free fatty acid levels cause metabolic inefficiency under ischemic conditions, as fatty acid oxidation requires more oxygen per ATP than glucose oxidation, increasing myocardial oxygen demand[22]. Lipotoxic intermediates (e.g., long-chain acyl-CoA, acylcarnitines) accumulate, causing mitochondrial dysfunction, oxidative stress, and apoptosis[23]. Free fatty acids are arrhythmogenic, shortening ventricular action potential duration and promoting malignant arrhythmias. They induce cardiac insulin resistance, impairing glucose utilization, a crucial oxygen-efficient fuel during ischemia. In addition, free fatty acids activate pro-inflammatory pathways (e.g., via TLR4), exacerbating endothelial dysfunction and inflammatory responses[24]. In the context of insulin resistance and metabolic syndrome, the heart becomes metabolically inflexible, relying excessively on fatty acid oxidation, which predisposes to energy depletion during stress. This leads to structural damage, including fibrosis, hypertrophy, and dysfunction, increasing susceptibility to MI and heart failure[25].
Elabscience® Quick Overview of Popular Products:
Table 1. Research Tools for Lipid Metabolism
|
Cat. No. |
Product Name |
|
E-BC-F065 |
Adipogenesis Fluorometric Assay Kit |
|
E-BC-F067 |
Fatty Acid Uptake Fluorometric Assay Kit |
|
E-BC-D011 |
3-Hydroxy-3-methylglutaryl-CoA Reductase (HMGR) Inhibitor Screening Kit |
|
E-BC-F032 |
Total Cholesterol and Cholesteryl Ester Fluorometric Assay Kit |
|
E-BC-F033 |
Triglyceride (TG) Fluorometric Assay Kit |
|
E-BC-F035 |
High-Density Lipoprotein Chlesterol (HDL-C) Fluorometric Assay Kit |
|
E-BC-F039 |
Free Fatty Acids (NEFA/FFA) Fluorometric Assay Kit |
|
E-BC-K004-M |
Free Cholesterol (FC) Colorimetric Assay Kit |
|
E-BC-K109-M |
Total Cholesterol (TC) Colorimetric Assay Kit |
|
E-BC-K205-M |
Low-Density Lipoprotein Cholesterol (LDL-C) Colorimetric Assay Kit |
|
E-BC-K221-M |
High-Density Lipoprotein Cholesterol (HDL-C) Colorimetric Assay Kit |
|
E-EL-M1363 |
Mouse LDL(Low Density Lipoprotein) ELISA Kit |
|
E-EL-H6279 |
Human LDL (Low Density Lipoprotein) ELISA Kit |
|
E-EL-H0160 |
Human LP-a(Lipoprotein A) ELISA Kit |
|
E-AB-F1291E |
APC Anti-Mouse CD36 Antibody[HM36] |
|
E-AB-F1164D |
PE Anti-Human CD36 Antibody[5-271] |
|
E-BC-K784-M |
Fatty Acid Oxidation (FAO) Colorimetric Assay Kit |
References:
[1] You, J., et al., Mapping the Plasma Metabolome to Human Health and Disease in 274,241 Adults. Nature Metabolism, 2025. 7(11):2366-2384.
[2] Wang, J., et al., Regulation of Lipoprotein Processing by GPNMB in Foamy Macrophages: Potential Therapeutic Targets for Atherosclerosis. Nature Communications, 2025. 16(1):10030.
[3] Gilliland, T.C., et al., Lipoprotein(a), Oxidized Phospholipids, and Coronary Artery Disease Severity and Outcomes. Journal of the American College of Cardiology, 2023. 81(18):1780-1792.
[4] Luciani, L., M. Pedrelli, and P. Parini, Modification of Lipoprotein Metabolism and Function Driving Atherogenesis in Diabetes. Atherosclerosis, 2024. 394:117545.
[5] Morita, S.-y., Metabolism and Modification of Apolipoprotein B-Containing Lipoproteins Involved in Dyslipidemia and Atherosclerosis. Biological & Pharmaceutical Bulletin, 2016. 39(1):1-24.
[6] Sakakura, K., et al., Pathophysiology of Atherosclerosis Plaque Progression. Heart, Lung and Circulation, 2013. 22(6):399-411.
[7] Shah, P.K., et al., Human Monocyte-Derived Macrophages Induce Collagen Breakdown in Fibrous Caps of Atherosclerotic Plaques: Potential Role of Matrix-Degrading Metalloproteinases and Implications for Plaque Rupture. Circulation, 1995. 92(6):1565-1569.
[8] Stanciulescu, L.A., A. Scafa-Udriste, and M. Dorobantu, Exploring the Association Between Low-Density Lipoprotein Subfractions and Major Adverse Cardiovascular Outcomes—A Comprehensive Review. International Journal of Molecular Sciences, 2023. 24(7):6669.
[9] Guo, B.-Q., et al., Comparative Efficacy of LDL-C-Lowering Therapies in First-Time vs. Recurrent Myocardial Infarction Prevention: A Meta-Analysis of Large-Scale Randomized Controlled Trials. European Journal of Preventive Cardiology, 2025.
[10] Sakamoto, K., et al., Effect of Ezetimibe Add-On Therapy Over 52 Weeks Extension Analysis of Prospective Randomized Trial (RESEARCH Study) in Type 2 Diabetes Subjects. Lipids in Health and Disease, 2017. 16(1):122.
[11] White, J.B., et al., Changes to the Serum Lipidome and Their Relation to Coronary Plaque in the First Six Months After Acute Myocardial Infarction. Atherosclerosis, 2025. 408:120421.
[12] Rong, J., et al., Serum Lipidomics Reveals Phosphatidylethanolamine and Phosphatidylcholine Disorders in Patients With Myocardial Infarction and Post-Myocardial Infarction-Heart Failure. Lipids in Health and Disease, 2023. 22(1):66.
[13] Liu, Y., et al., Identification of Regulator Gene and Pathway in Myocardial Ischemia-Reperfusion Injury: A Bioinformatics and Biological Validation Study. Hereditas, 2025. 162(1):35.
[14] Pilz, S., et al., Elevated Plasma Free Fatty Acids Predict Sudden Cardiac Death: A 6.85-Year Follow-Up of 3315 Patients After Coronary Angiography. European Heart Journal, 2007. 28(22):2763-2769.
[15] Sun, Y., et al., Assessment of the Association Between Cardiac Metabolic Markers and Carotid Atherosclerosis, and the Role of Insulin Resistance. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy, 2025. 18:2899-2911.
[16] Kosmas, C.E., et al., Insulin Resistance and Cardiovascular Disease. Journal of International Medical Research, 2023. 51(3):3000605231164548.
[17] Grundy, S.M., Pre-Diabetes, Metabolic Syndrome, and Cardiovascular Risk. Journal of the American College of Cardiology, 2012. 59(7):635-643.
[18] Hotamisligil, G.S., Inflammation, Metaflammation and Immunometabolic Disorders. Nature, 2017. 542(7640):177-185.
[19] Sattar, N., et al., Metabolic Syndrome With and Without C-Reactive Protein as a Predictor of Coronary Heart Disease and Diabetes in the West of Scotland Coronary Prevention Study. Circulation, 2003. 108(4):414-419.
[20] Taskinen, M.R., and J. Borén, New Insights Into the Pathophysiology of Dyslipidemia in Type 2 Diabetes. Atherosclerosis, 2015. 239(2):483-495.
[21] Marso, S.P., et al., Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. New England Journal of Medicine, 2016. 375(4):311-322.
[22] Lopaschuk, G.D., et al., Myocardial Fatty Acid Metabolism in Health and Disease. Physiological Reviews, 2010. 90(1):207-258.
[23] Goldberg, I.J., C.M. Trent, and P.C. Schulze, Lipid Metabolism and Toxicity in the Heart. Cell Metabolism, 2012. 15(6):805-812.
[24] Shi, H., et al., TLR4 Links Innate Immunity and Fatty Acid–Induced Insulin Resistance. Journal of Clinical Investigation, 2006. 116(11):3015-3025.
[25] Lopaschuk, G.D., et al., Cardiac Energy Metabolism in Heart Failure. Circulation Research, 2021. 128(10):1487-1513.

