T cell-mediated inflammation is a central driver of metabolic dysfunction and tissue injury in type 2 diabetes (T2D). This process is characterized by a systemic imbalance, favoring pro-inflammatory T helper (Th) 1 and Th17 cells over regulatory T cells (Tregs). In pancreatic islets, Th17-derived IL-17 directly impairs β-cell function and amplifies local inflammation, accelerating β-cell failure. Within obese adipose tissue, a pathological crosstalk between activated T cells and macrophages creates a self-sustaining cycle of cytokine production that directly induces insulin resistance in adipocytes. Furthermore, dysregulated checkpoint molecules like PD-1 and a milieu of T cell-derived cytokines promote inhibitory serine phosphorylation of insulin receptor substrates (IRS), crippling insulin signaling across metabolic tissues.
In this context, we highlight the pivotal role of adaptive immune dysregulation as a critical and targetable axis in the pathophysiology of T2D. Correcting the Th17/Treg imbalance, disrupting the adipose immune cycle, and modulating specific T cell metabolic pathways represent promising therapeutic strategies to preserve β-cell mass, restore insulin sensitivity, and halt disease progression.
Table of Contents
1. Contribution of Th17 cells to pancreatic islet inflammation
2. Regulatory T cell (Treg) frequency and function in insulin-resistant liver
3. Crosstalk between T cells and macrophages in adipose tissue inflammation
4. Impact of high-fat diet on T cell-mediated cytokine profiles in metabolic tissues
5. Effect of checkpoint molecules (PD-1, CTLA-4) on T cell activity in metabolic inflammation
6. Assessment of T cell-derived cytokine impact on insulin receptor substrate phosphorylation
01 Contribution of Th17 cells to pancreatic islet inflammation
Pancreatic islet inflammation (insulitis) is a key driver of β-cell dysfunction in type 2 diabetes (T2D), with T helper 17 (Th17) cells serving as pivotal mediators. A systemic and islet-localized imbalance favoring pro-inflammatory Th17 cells over regulatory T cells (Tregs) establishes a deleterious inflammatory milieus[1].
The primary effector mechanism involves interleukin-17 (IL-17), the signature cytokine of Th17 cells[1]. IL-17 binding to its receptor activates NF-κB and JNK pathways within β-cells and resident immune cells. This activation induces the production of pro-inflammatory cytokines (e.g., TNF-α, IL-1β) and chemokines, which recruit additional immune cells and amplify local inflammation. Crucially, IL-17 also directly impairs β-cell function and insulin secretion[2]. The resulting inflammatory cascade synergistically promotes β-cell apoptosis and endoplasmic reticulum stress[3, 4].
Clinical evidence supports this pathway, with IL-17 detected in T2D islets and circulating Th17 frequency correlating with poor β-cell function and glycemic control[5]. Therefore, targeting the Th17 axis or correcting the Th17/Treg imbalance represents a promising therapeutic strategy for preserving β-cell mass and function in T2D.
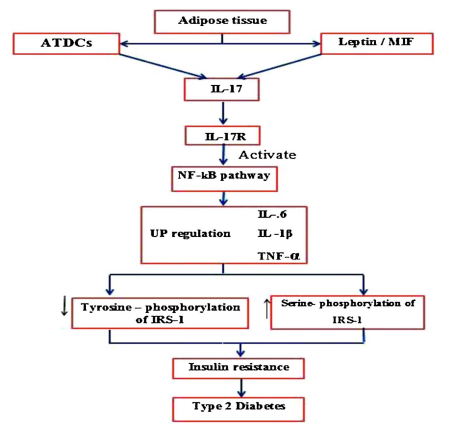
Fig. 1 Mechanism and the role of IL-17 in development of T2D[1]. Adipose tissue dendritic cells (ATDCs) promote inflammation by producing IL-17, a process also stimulated by leptin and migration inhibitory factor. IL-17 activates the NF-κB pathway, increasing pro-inflammatory cytokines like IL-1β and TNF-α. TNF-α directly inhibits insulin signaling by inducing serine phosphorylation of IRS-1, leading to insulin resistance and type 2 diabetes.
02 Regulatory T cell (Treg) frequency and function in insulin-resistant liver
The liver is central to systemic metabolic homeostasis, and its immune environment is critically disrupted in insulin resistance and T2D. Tregs, essential for maintaining immune tolerance, play a complex and context-dependent role in metabolic liver disease.
In early metabolic stress, a deficiency in hepatic Treg frequency or function contributes to inflammation, exacerbating hepatic steatosis and insulin resistance[6]. This aligns with clinical observations of diminished or dysfunctional Tregs in T2D, which perpetuates a pro-inflammatory state characterized by elevated cytokines like TNF-α and IL-6 that impair insulin signaling.
Paradoxically, in chronic conditions such as non-alcoholic steatohepatitis (NASH), sustained activation can render the hepatic Treg response maladaptive. The enrichment of a specific Treg subset producing amphiregulin (AREG) is key. While beneficial in acute repair, chronic exposure to Treg-derived AREG activates hepatic stellate cells via epidermal growth factor receptor (EGFR) signaling, driving liver fibrosis and worsening metabolic outcomes[7]. This reveals a functional duality: Tregs suppress initial inflammation but may promote fibrotic remodeling when chronically engaged[8].
The molecular pathways governing Tregs in the insulin-resistant liver involve intricate intercellular crosstalk. For instance, signaling in hepatic macrophages can modulate Tregs via mechanisms like the exosomal miR-142a-3p/TGFβ receptor 1 axis[8]. Thus, the net impact of Tregs is determined not merely by their quantity but by their functional polarization and the local microenvironment. This underscores the therapeutic challenge of selectively preserving Treg-mediated anti-inflammatory protection while inhibiting pro-fibrotic activities.
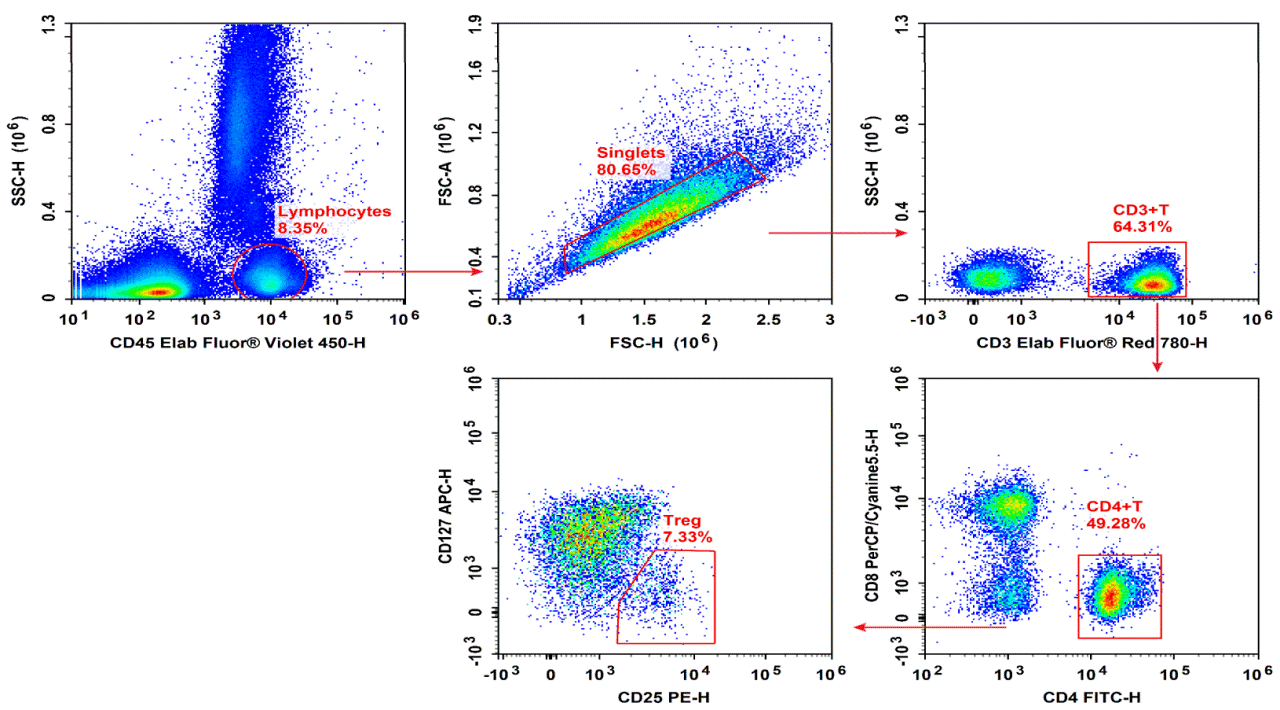
Fig. 2 Normal human peripheral blood cells are stained with EV450 Anti-Human CD45, ER780 Anti-Human CD3, FITC Anti-Human CD4, PERCP-CY5.5 Anti-Human CD8, APC Anti-Human CD127 and PE Anti-Human CD25, followed by analysis via flow cytometry. (The data are provided by Elabscience)
03 Crosstalk between T cells and macrophages in adipose tissue inflammation
Adipose tissue inflammation in obesity is driven by a self-sustaining cycle of reciprocal activation between T cells and macrophages, constituting a central mechanism of metabolic dysfunction[9].
T cell activation within the obese adipose niche initiates this process. Antigen-presenting cells, including macrophages and dendritic cells, activate tissue-resident T cells and promote the differentiation of pro-inflammatory Th1 cells that produce interferon-γ (IFN-γ). Adipose tissue macrophages (ATMs) are functional antigen-presenting cells that promote the proliferation of IFN‑γ‑producing CD4+ T cells in adipose tissue[10].
This activation triggers the core inflammatory loop. Th1-derived IFN-γ recruits monocytes and drives their polarization into pro-inflammatory M1 macrophages[11]. These M1 cells, in turn, perpetuate T cell activation by presenting antigens and providing co-stimulation, creating a feed-forward cycle that amplifies cytokine production (e.g., IL-6) and sustains chronic inflammation[12].
The overall inflammatory tone is regulated by the balance of T cell subsets. While Th1 cells propel the vicious cycle, regulatory T cells (Tregs) and Th2 cells secrete anti-inflammatory cytokines (IL-10, IL-4, IL-13) that support resolution via M2 macrophages[13]. Obesity disrupts this balance, favoring the pro-inflammatory axis sustained by the pathological crosstalk between T cells and macrophages[14].
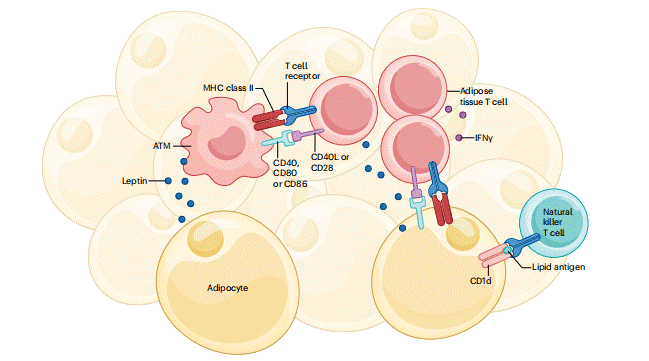
Fig. 3 Integration of adipocyte, ATM and adipose tissue T cell signalling during obesity[9]. During obesity, adipocytes and ATMs activate adipose tissue T cells by presenting antigens and providing co-stimulatory signals, triggering IFN-γ production. Leptin from adipocytes further shapes this pro-inflammatory immune environment. These interactions promote the accumulation of inflammatory immune cells within fat tissue, contributing to systemic inflammation and glucose intolerance.
04 Impact of high-fat diet on T cell-mediated cytokine profiles in metabolic tissues
High-fat diet (HFD) consumption is a major environmental driver that reshapes the T cell landscape, exacerbating pancreatic islet inflammation and β‑cell dysfunction. HFD induces a systemic and islet-localized shift toward pro‑inflammatory Th1 and Th17 phenotypes while reducing Tregs, creating a cytokine milieu dominated by IFN‑γ and IL‑17.
Mechanistically, HFD exerts its effects through multiple interconnected pathways:
Metabolic Reprogramming of T Cells: Elevated saturated fatty acids (e.g., palmitic acid) and chronic hyperglycemia modulate T cell function. Th17 differentiation and function are particularly sensitive to lipid metabolism, requiring fatty acid uptake. Pharmacological blockade of carnitine Palmitoyltransferase 1A(CPT1A), a key enzyme in fatty acid oxidation, inhibits Th17‑associated cytokine production[15]. Palmitic acid can upregulate surface receptors like SLAMF3 via the STAT5‑PI3K/Akt pathway, driving chronic inflammatory T cell profiles[16].
Key Signaling Mediators: HFD enhances p38α activity in T cells. Genetic deletion of p38α specifically in T cells protects against HFD‑induced insulin resistance and islet inflammation, underscoring its critical role[17].
Disruption of Treg Homeostasis: Concurrently, HFD impairs Treg function by disturbing cholesterol metabolism and clonal expansion of protective subsets, further tilting the Th17/Treg balance toward inflammation.
Sustained Inflammatory Output: Despite HFD‑induced reductions in overall T cell glycolytic and mitochondrial metabolism, inflammatory cytokine production (including IL‑17) is paradoxically sustained. This perpetuates β‑cell injury and insulin resistance[15].Within pancreatic islets, infiltrating and locally activated Th17 cells secrete IL‑17, which activates NF‑κB and JNK signaling in β‑cells. This direct action, combined with the recruitment and activation of other immune cells, amplifies local inflammation and impairs β‑cell function. Thus, HFD‑driven immunometabolic reprogramming of T cells constitutes a critical link between dietary fat and the progression of islet inflammation in T2D.
%20cells_.png)
Fig. 4 Peripheral blood mononuclear cells (PBMCs) were cultured with Cell Stimulation MIX and Protein Transport Inhibitor MIX for 5 hours, then were harvested, fixed, and permeabilized, followed by staining with PerCP/Cyanine5.5 Anti-Human CD3, Elab Fluor® 488 Anti-Human CD4 and PE Anti-Human IL-17A for analysis of the proportion of Th17 cells and their functional capacity to secrete the effector cytokine IL-17A. Th17 cells were defined by the phenotype CD3+CD4+IL-17A+. (The data are provided by Elabscience)
05 Effect of checkpoint molecules (PD-1, CTLA-4) on T cell activity in metabolic inflammation
Immune checkpoint molecules programmed death-1(PD-1) and CTLA-4 are critical negative regulators of T cell activation. The canonical two signal model (CD3/CD28 T cell activation) is potently inhibited by these checkpoints, which maintain immune homeostasis and prevent autoimmunity. Dysregulation of PD 1 and CTLA 4 pathways is increasingly recognized as a contributor to persistent T cell activation in metabolic inflammation, including type 2 diabetes (T2D) and associated liver disease.
PD 1 and CTLA 4 employ distinct metabolic reprogramming mechanisms to suppress T cell activity. PD 1 ligation antagonizes PI3K/AKT signaling, thereby inhibiting glycolysis and promoting fatty acid oxidation (FAO), which blocks effector T cell differentiation[18]. In contrast, CTLA 4 primarily suppresses glycolysis without augmenting FAO, sustaining a metabolic profile similar to non activated cells. These distinct mechanisms underscore how checkpoint molecules constrain CD3/CD28 driven T cell activation under chronic metabolic stress.
In T2D, downregulation of PD 1 on immune cells may unleash T cell activity, exacerbating tissue inflammation. Conversely, partially dysfunctional PD 1⁺CD4⁺ T cells have been identified as inter organ mediators of insulin resistance. The clinical relevance of these pathways is highlighted by immune checkpoint inhibitors (ICIs), including anti PD 1 antibody and anti CTLA 4 antibody. While effective in oncology, these agents cause immune related adverse events such as autoimmune diabetes, with combination therapy increasing the risk of checkpoint inhibitor induced diabetes mellitus[19, 20]. Moreover, PD‑1 signaling in adipose tissue group 2 innate lymphoid cells (ILC2s) also contributes to metabolic regulation, as PD‑1 blockade can ameliorate insulin resistance in obese mice[21]. Thus, PD 1 and CTLA 4 differentially regulate T cell metabolism and CD3/CD28 dependent activation, with therapeutic manipulation carrying significant implications for metabolic inflammation.
06 Assessment of T cell-derived cytokine impact on insulin receptor substrate phosphorylation
Cytokines derived from activated T cells directly modulate insulin receptor substrate (IRS) phosphorylation, establishing a critical link between immune activation and metabolic dysfunction. IRS-1 and IRS-2 are essential docking proteins downstream of the insulin receptor; their tyrosine phosphorylation is required for PI3K/AKT activation and glucose homeostasis.
The impact of T cell cytokines on IRS phosphorylation is dichotomous:
Promotion of Tyrosine Phosphorylation: A subset of interleukins, including IL-2, IL-9, and IL-15, can rapidly stimulate tyrosine phosphorylation of IRS-1/2 in lymphoid cells via associated Janus kinases (JAKs), with JAK1 and JAK3 directly phosphorylating IRS-2[22]. This represents a potential positive regulatory axis within the immune system.
Induction of Inhibitory Serine Phosphorylation: In contrast, pro-inflammatory cytokines elevated during T cell-mediated inflammation (e.g. IL-1β, IL-17) promote inhibitory serine phosphorylation of IRS-1 (e.g., on Ser307/312). These cytokines activate stress kinases (JNK, IKKβ) and other pathways, leading to IRS-1 serine phosphorylation[23]. This modification uncouples IRS from the insulin receptor, impairing downstream PI3K/Akt signaling, GLUT4 translocation, and glucose uptake.
Within the milieu of T cell-mediated inflammation in metabolic tissues, the balance shifts decisively toward the action of pro-inflammatory cytokines. Thus, T cell-derived cytokines can disrupt IRS-dependent insulin signaling in parenchymal cells, directly linking inflammation to insulin resistance. Concurrently, certain cytokines may engage IRS pathways within T cells themselves, suggesting an integrated immunometabolic crosstalk[24]. Targeting these cytokine–IRS axes may offer therapeutic opportunities to restore insulin sensitivity in T2D.
Quick Overview of Popular Products:
Table 1. Reagents for T cell-mediated inflammation and type 2 diabetes research
|
Cat. No. |
Product Name |
|
E-BC-K784-M |
Fatty Acid Oxidation (FAO) Colorimetric Assay Kit |
|
E-AB-F1173D |
PE Anti-Human IL-17A Antibody[BL168] |
|
E-EL-H5812 |
Human IL-17A (Interleukin 17A) ELISA Kit |
|
E-EL-H0109 |
Human TNF-α (Tumor Necrosis Factor Alpha) ELISA Kit |
|
E-EL-R2856 |
Rat TNF-α (Tumor Necrosis Factor Alpha) ELISA Kit |
|
E-EL-H2665 |
Human INS (Insulin) ELISA Kit |
|
E-EL-R2466 |
Rat INS (Insulin) ELISA Kit |
|
E-OSEL-H0019 |
QuicKey Pro Human INS (Insulin) ELISA Kit |
|
XJH002 |
Human Th17 Flow Cytometry Staining Kit |
|
MIH001A |
Human CD3/CD28 T Cell Activation Beads |
|
E-AB-F1131H |
PE/Cyanine7 Anti-Mouse CD279/PD-1 Antibody[29F.1A12] |
|
XJM004 |
RAW 264.7 Polarized M1 Macrophage Induction and Identification Kit |
|
E-EL-H0108 |
Human IFN-γ (Interferon Gamma) ELISA Kit |
|
E-AB-F1196D |
PE Anti-Human IFN-γ Antibody[B27] |
|
E-BC-F067 |
Succinate Dehydrogenase (SDH) Activity Assay Kit |
|
E-CK-A441 |
Fatty Acid Uptake Fluorometric Assay Kit |
References:
[1] Abdel-Moneim, A., H.H. Bakery, and G. Allam, The Potential Pathogenic Role of IL-17/Th17 Cells in Both Type 1 and Type 2 Diabetes Mellitus. Biomedicine & Pharmacotherapy, 2018. 101: 287-292.
[2] Chen, J., et al., Inflammatory Signaling Pathways in Pancreatic β-Cell: New Insights into Type 2 Diabetes Pathogenesis. Pharmacological Research, 2025. 216:107776.
[3] Vasilev, G., et al., T Helper 17 Cells and Interleukin-17 Immunity in Type 1 Diabetes: From Pathophysiology to Targeted Immunotherapies. World Journal of Diabetes, 2025. 16(4):99936.
[4] Arif, S., et al., Peripheral and Islet Interleukin-17 Pathway Activation Characterizes Human Autoimmune Diabetes and Promotes Cytokine-Mediated β-Cell Death. Diabetes, 2011. 60(8):2112-2119.
[5] Rajendran, S., et al., IL-17 Is Expressed on Beta and Alpha Cells of Donors with Type 1 and Type 2 Diabetes. Journal of Autoimmunity, 2021. 123:102708.
[6] Zhang, M., et al., Macrophage Notch1 Signaling Modulates Regulatory T Cells via the TGFB Axis in Early MASLD. JHEP Reports, 2025. 7(1):101242.
[7] Savage, T.M., et al., Amphiregulin from Regulatory T Cells Promotes Liver Fibrosis and Insulin Resistance in Non-Alcoholic Steatohepatitis. Immunity, 2024. 57(2):303-318.e6.
[8] Ajith, A., et al., Immune Regulation and Therapeutic Application of T Regulatory Cells in Liver Diseases. Frontiers in Immunology, 2024. 15:1371089.
[9] Jacks, R.D. and C.N. Lumeng, Macrophage and T Cell Networks in Adipose Tissue. Nature Reviews Endocrinology, 2024. 20(1):50-61.
[10] Morris, D.L., et al., Adipose Tissue Macrophages Function as Antigen-Presenting Cells and Regulate Adipose Tissue CD4+ T Cells in Mice. Diabetes, 2013. 62(8): 2762-2772.
[11] Wang, Q. and H. Wu, T Cells in Adipose Tissue: Critical Players in Immunometabolism. Frontiers in Immunology, 2018. 9:2509.
[12] Cho, K.W., et al., An MHC II-Dependent Activation Loop between Adipose Tissue Macrophages and CD4+ T Cells Controls Obesity-Induced Inflammation. Cell Reports, 2014. 9(2):605-617.
[13] Elkins, C., et al., Obesity Reshapes Regulatory T Cells in the Visceral Adipose Tissue by Disrupting Cellular Cholesterol Homeostasis. Science Immunology, 2025. 10(103):eadl4909.
[14] Deng, T., et al., Adipocyte Adaptive Immunity Mediates Diet-Induced Adipose Inflammation and Insulin Resistance by Decreasing Adipose Treg Cells. Nature Communications, 2018. 8(1):15725.
[15] Nicholas, D.A., et al., Fatty Acid Metabolites Combine with Reduced β Oxidation to Activate Th17 Inflammation in Human Type 2 Diabetes. Cell Metabolism, 2019. 30(3):447-461.e5.
[16] Zhou, T., et al., Upregulation of SLAMF3 on Human T Cells Is Induced by Palmitic Acid through the STAT5-PI3K/Akt Pathway and Features the Chronic Inflammatory Profiles of Type 2 Diabetes. Cell Death & Disease, 2019. 10(8): 559.
[17] Meng, D., et al., p38α Deficiency in T Cells Ameliorates Diet-Induced Obesity, Insulin Resistance, and Adipose Tissue Senescence. Diabetes, 2022. 71(6):1205-1217.
[18] Boussiotis, V.A. and N. Patsoukis, Effects of PD-1 Signaling on Immunometabolic Reprogramming. Immunometabolism, 2022. 4(2).
[19] Mourad, D., et al., Immune Checkpoint Inhibitor-Induced Diabetes Mellitus: Potential Role of T Cells in the Underlying Mechanism. International Journal of Molecular Sciences, 2021. 22(4).
[20] Perdigoto, A.L., et al., Immune Cells and Their Inflammatory Mediators Modify β Cells and Cause Checkpoint Inhibitor-Induced Diabetes. JCI Insight, 2022. 7(17).
[21] Sun, J., et al., Metabolic Regulator LKB1 Controls Adipose Tissue ILC2 PD-1 Expression and Mitochondrial Homeostasis to Prevent Insulin Resistance. Immunity, 2024. 57(6):1289-1305.e9.
[22] Johnston, J.A., et al., Interleukins 2, 4, 7, and 15 Stimulate Tyrosine Phosphorylation of Insulin Receptor Substrates 1 and 2 in T Cells. Potential Role of JAK Kinases. Journal of Biological Chemistry, 1995. 270(48): 28527-28530.
[23] de Luca, C. and J.M. Olefsky, Inflammation and Insulin Resistance. FEBS Letters, 2008. 582(1):97-105.
[24] Hameed, I., et al., Type 2 Diabetes Mellitus: From a Metabolic Disorder to an Inflammatory Condition. World Journal of Diabetes, 2015. 6(4): 598-612.

