Researchers fty have fund that PXR is crucial for protecting against AKI by targeting the AKR1B7/mitochondrial metabolism axis. Praise to research team of Professor Aihua Zhanh and Zhanjun Jia, Elabscience is honored to support this significant study by providing ELISA products.
Fundamental Information
Title: Nuclear receptor PXR targets AKR1B7 to protect mitochondrial metabolism and renal function in AKI
Journal: Science Translational Medicine
Impact Factor: 17.161
Institution of the first author: Nanjing Medical University
Institution of the corresponding author: Nanjing Medical University
Elabscience’s Products Cited:
|
Cat. No |
Application |
Detection target |
Species |
Tested sample |
|
ELISA |
Mouse |
Serum |
||
|
ELISA |
Mouse |
Serum |


Background of Research
Acute kidney injury (AKI) is a life-threatening illness which affects 13.5 million patients and about 1.7 million people die per year globally due to AKI. Unavailability of satisfactory therapies to treat established AKI critically restrict clinical efforts to recovery in patients with AKI. Therefore, new strategies that minimize AKI are urgently needed.
Normal kidney function depends on different cell types working in concert, which involves many energy-intensive processes . Mitochondria in renal proximal tubular epithelial cells synthesize adenosine 5′-triphosphate (ATP) through electron transport and oxidative phosphorylation (OXPHOS) in conjunction with the oxidation of metabolites by the tricarboxylic acid cycle and catabolism of fatty acids by -oxidation. Energy homeostasis could collapse due to interruption of mitochondrial metabolism resulting in AKI. As energy is metabolized by mitochondria, a potential approach could be developed to alleviate AKI by targeting mitochondrial metabolism.
Nuclear receptors control a large variety of metabolic processes, including kidney lipid metabolism, drug clearance, inflammation, fibrosis, cell differentiation, and oxidative stress. The pregnane X receptor (PXR) is a member of the nuclear receptor (NR) protein family (classified as Nr1i2) and is mainly expressed in organs with tubular epithelial structure, such as the kidney, liver, and gut. PXR can be widely involved in the body’s material and energy metabolism by transcriptionally regulating the expression of downstream target genes. In addition, PXR regulate glycolipid metabolism, inflammation, and atherosclerosis. However, dysregulation of PXR occurs in CKD, but no functional examination of its role in CKD pathology has been conducted. PXR activation by its ligands could increase human diseases such as cholestatic liver diseases. Till now, no involvement of PXR in AKI is reported.
Quantitative proteomic analysis (iTRAQ) was conducted before defining the role of PXR in AKI to assess differential protein expression in kidneys from wild-type and PXR−/− rats. The expression of Aldo-keto reductase family 1, member B7 (AKR1B7) was markedly lower in kidneys of PXR−/−rats than in those of wild-type controls, suggesting that Akr1b7 might be a target gene of PXR. Previous studies showed that AKR1B7 is expressed in mouse kidney, steroidogenic tissues, intestine and liver and has been suggested to play an important role in the detoxification of lipid peroxidation by-products and in lipid metabolism and a major regulator of white adipose tissue development.
Hence, PXR-controlled AKR1B7 may play an important role in maintaining metabolic homeostasis and organ function in various contexts. However, this pathway has not been investigated in kidney disease.
Purpose of Research
To investigate the role of PXR in pathogenesis of AKI and underlying mechanism(s) involved in its function.
Experimental Design
In this study, human kidney tissues, AKI models in rats and mice, and in vitro models of renal tubular cell injury were used to determine the role of PXR and the potential mechanisms by demonstrating the importance of PXR/AKR1B7 signaling in protecting against AKI and improving mitochondrial metabolism (OXPHOS and -oxidation).
Research Findings
1. The down-regulation of PXR in kidneys was associated with AKI
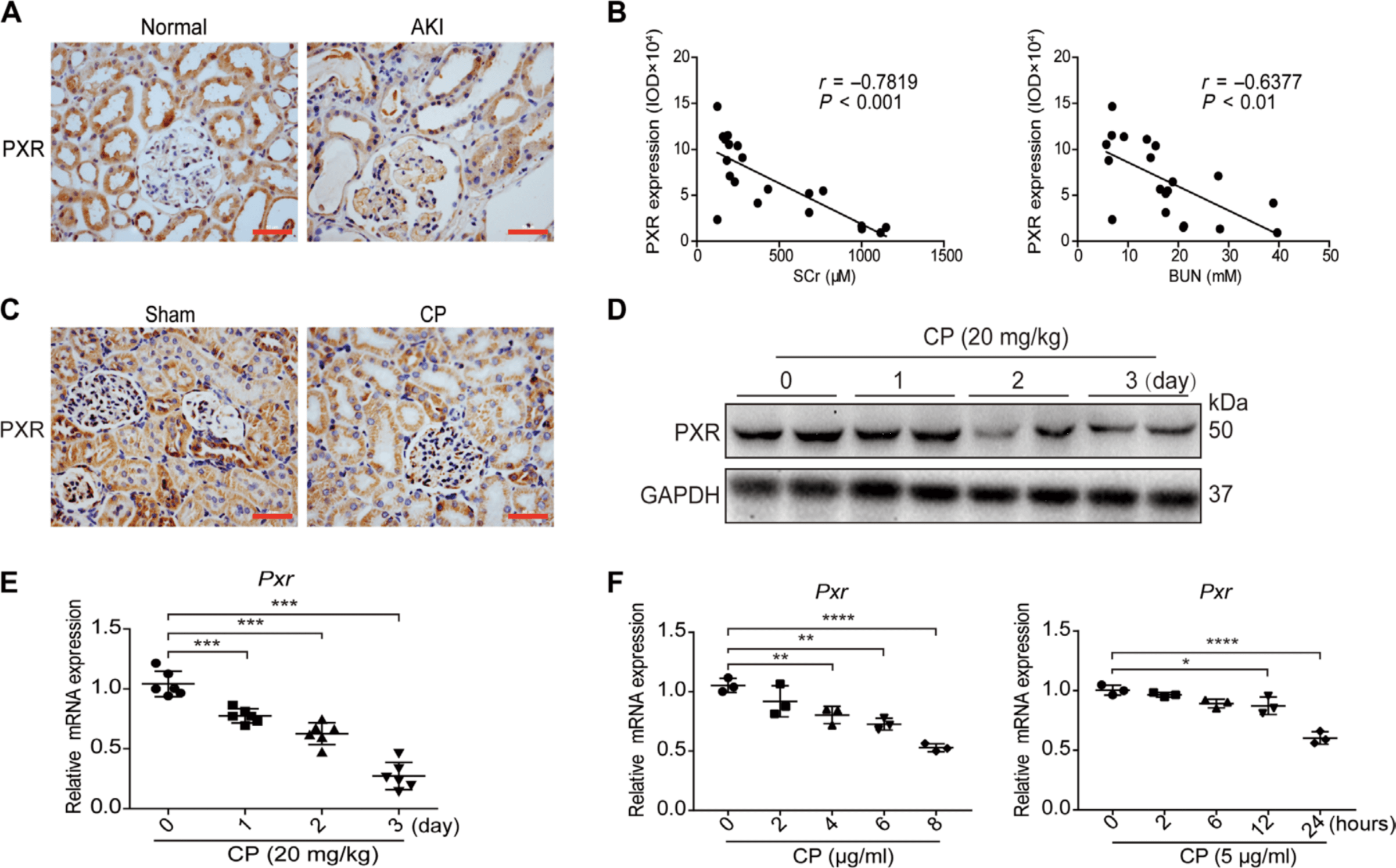
· PXR expression reduced in kidneys from patients with AKI
· PXR expression was negatively correlated with the peak concentration of blood urea nitrogen (BUN) and serum creatinine (SCr) in renal tubular cells and demonstrate that PXR down-relulation is associated with the pathogenesis of acute renal tubular injury
· PXR expression in the kidneys of cisplatin-treated mice noticeably decreased in a time-dependent manner
2. PXR deficiency aggravated cisplatin-induced AKI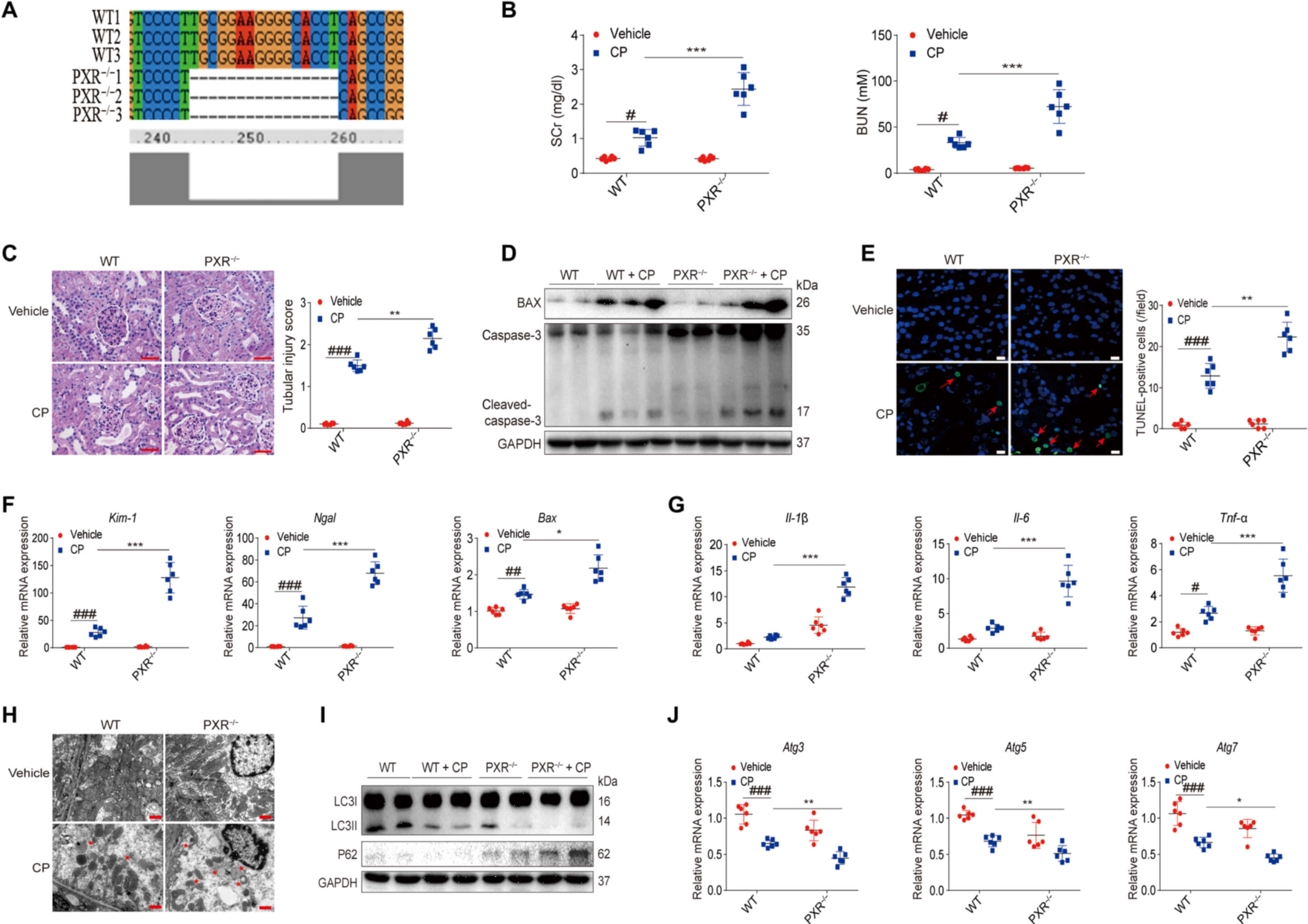
· The SCr and BUN concentrations in cisplatin treated PXR−/− rats were markedly higher
· Worse tubular injury was found in PXR−/− rats after cisplatin treatment
· The expressions of BAX and cleaved caspase-3 and the number of TUNEL–positive cells in the kidneys of cisplatin-treated PXR−/− rats were higher
· mRNA expressions of the tubular injury markers kidney injury molecule 1 (Kim-1) and neutrophil gelatinase–associated lipocalin (Ngal), inflammatory factors (Il-1b, Il-6, and Tnf-a), and Bax in the kidneys of cisplatin-treated PXR−/− rats were higher
· Swollen mitochondria and disrupted cristae were evident in the kidneys of cisplatin-treated wild-type rats, and these morphological changes worsen in cisplatin-treated PXR−/− rats.
· PXR inactivation impairs mitophagy and mitochondrial function that are important in AKI
3.Activation of PXR by PCN attenuated cisplatin-induced AKI
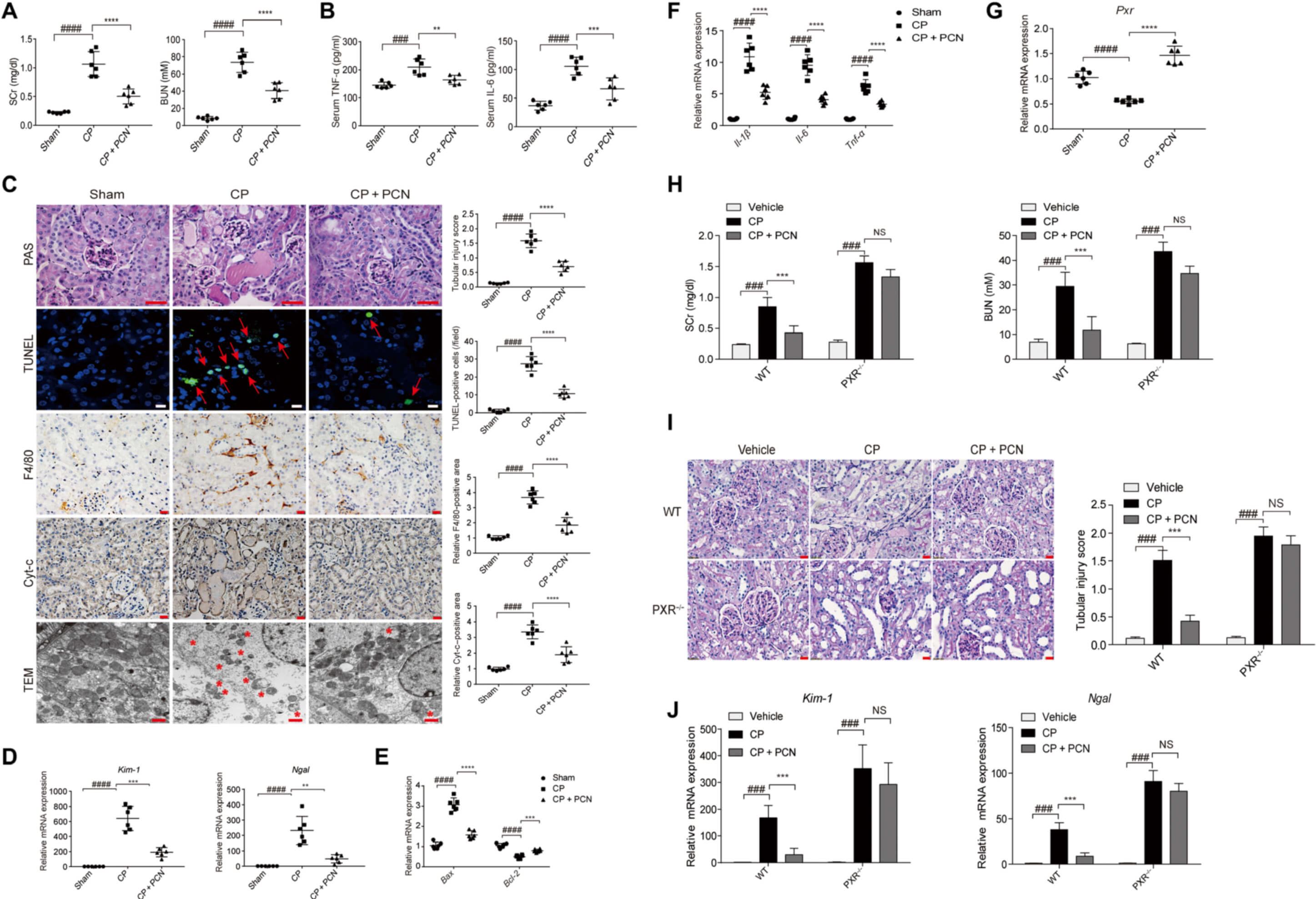
· PCN reduced the SCr and BUN concentrations in cisplatin-treated mice
· Increased circulating protein concentrations of interleukin-6 (IL-6) and tumor necrosis factor– (TNF-) in AKI mice were reduced upon the addition of PCN
· PCN mitigated tubular injury along with a decrease in the release of Cytochrome c (Cyt-c) from mitochondria after cisplatin treatment and reduced the number of TUNEL-positive cells and F4/80-positive cells
· PCN treatment lowered the mRNA expression of tubular injury markers (Kim-1 and Ngal), apoptosis-associated proteins (Bax and Bcl-2), and inflammatory factors (Il-1, Il-6, and Tnf-) in the kidneys
· Pxr mRNA expression was also increased upon exposure to PCN
4.Overexpression of PXR in kidneys protected against cisplatin-induced AKI
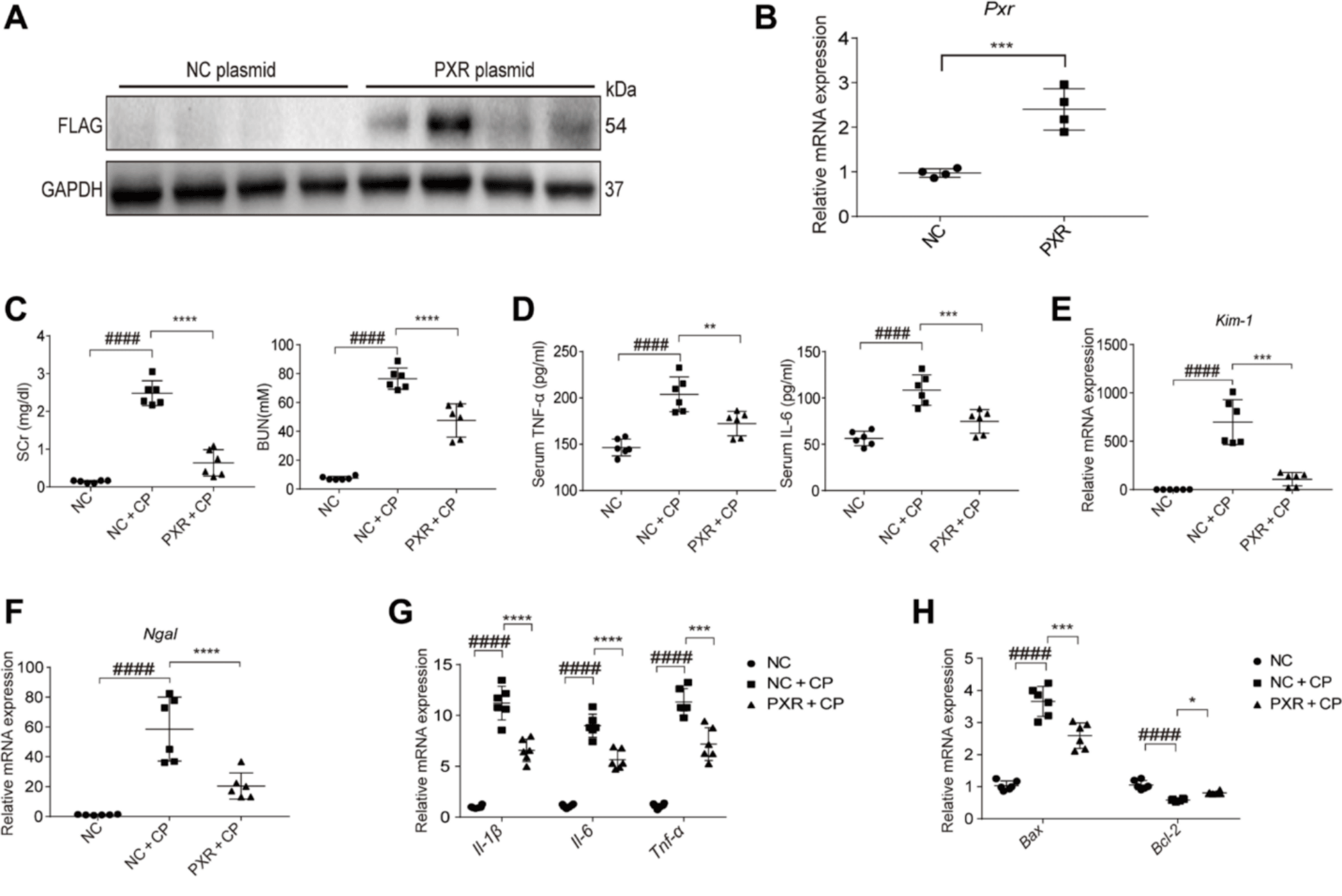
· mRNA and protein expression of FLAG-tagged PXR markedly increased in the kidneys 36 hours after injection of the PXR plasmids. This gene delivery approach efficiently
overexpresses
· PXR in the kidneys
· PXR plasmids markedly improved renal function in AKI mice
· the tubular injury scores, Cyt-c release from mitochondria, altered mitochondrial morphology
5.PCN treatment or PXR overexpression attenuated cisplatin-induced mitochondrial dysfunction in vitro
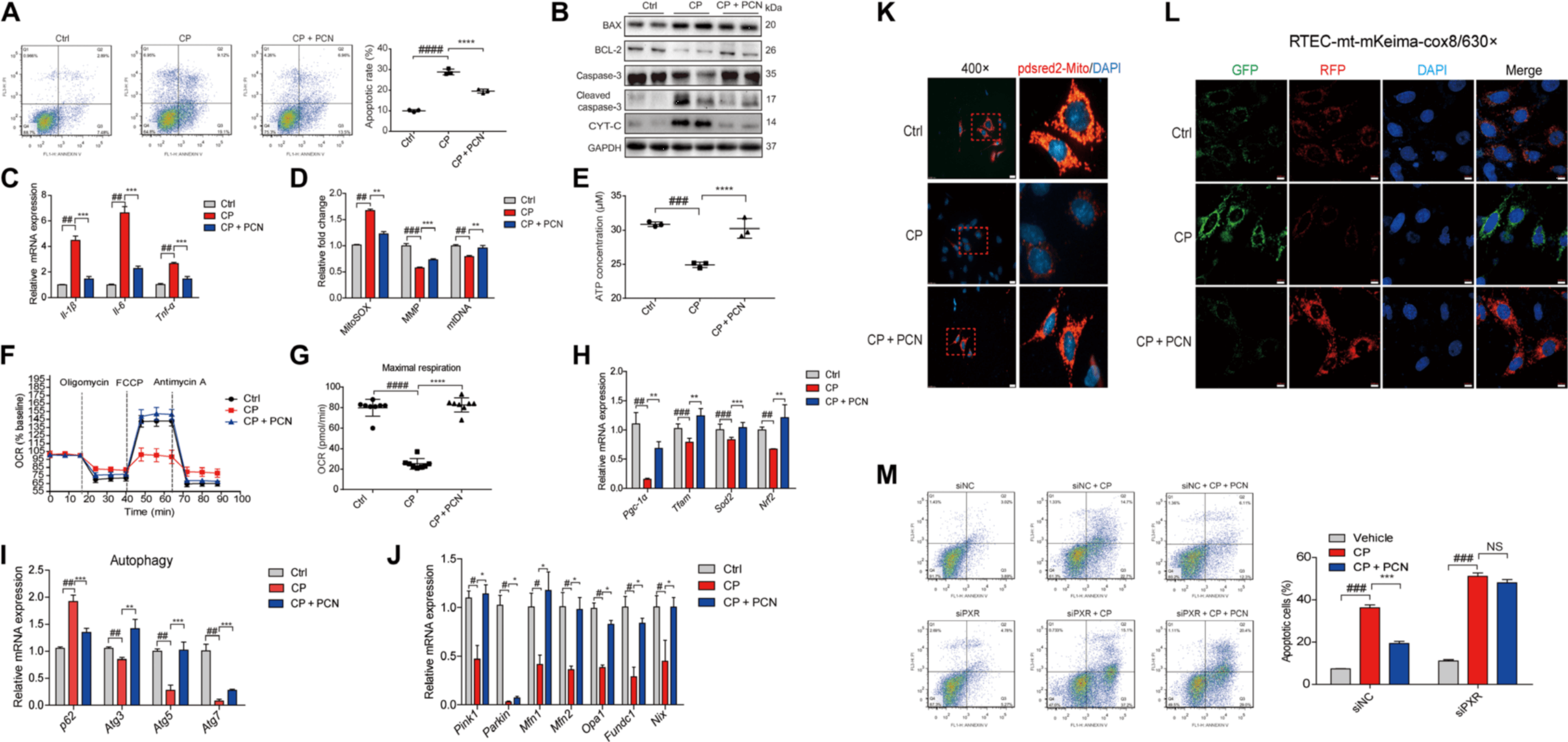
· PCN treatment prevented cisplatin-induced injury in RTECs and primary renal tubular cells, as shown by reduced cellular apoptosis; decreased expression of BAX, cleaved caspase-3, and Cyt-c; and restored expression of B-cell lymphoma-2 (BCL-2)
· PCN treatment reduced cisplatin-induced accumulation of mitochondrial ROS
· PCN treatment increased the expressions of Pgc-1a, Tfam, Sod2, and Nrf2
· PCN activated mitophagy, as seen by the increased expressions of Atg3, Atg5, and Atg7 and decreased expression of p62
· the intensity of red fluorescence decreased in cisplatin treated RTECs and recovered upon PCN treatment
· silencing of PXR abrogated the inhibition of cisplatin-induced apoptosis and inflammatory cytokine production by PCN
· PXR overexpression also improved mitochondrial homeostasis and mitophagy
6.Akr1b7 was the transcriptional target of PXR in kidneys and protected against CP-induced AKI in vivo.
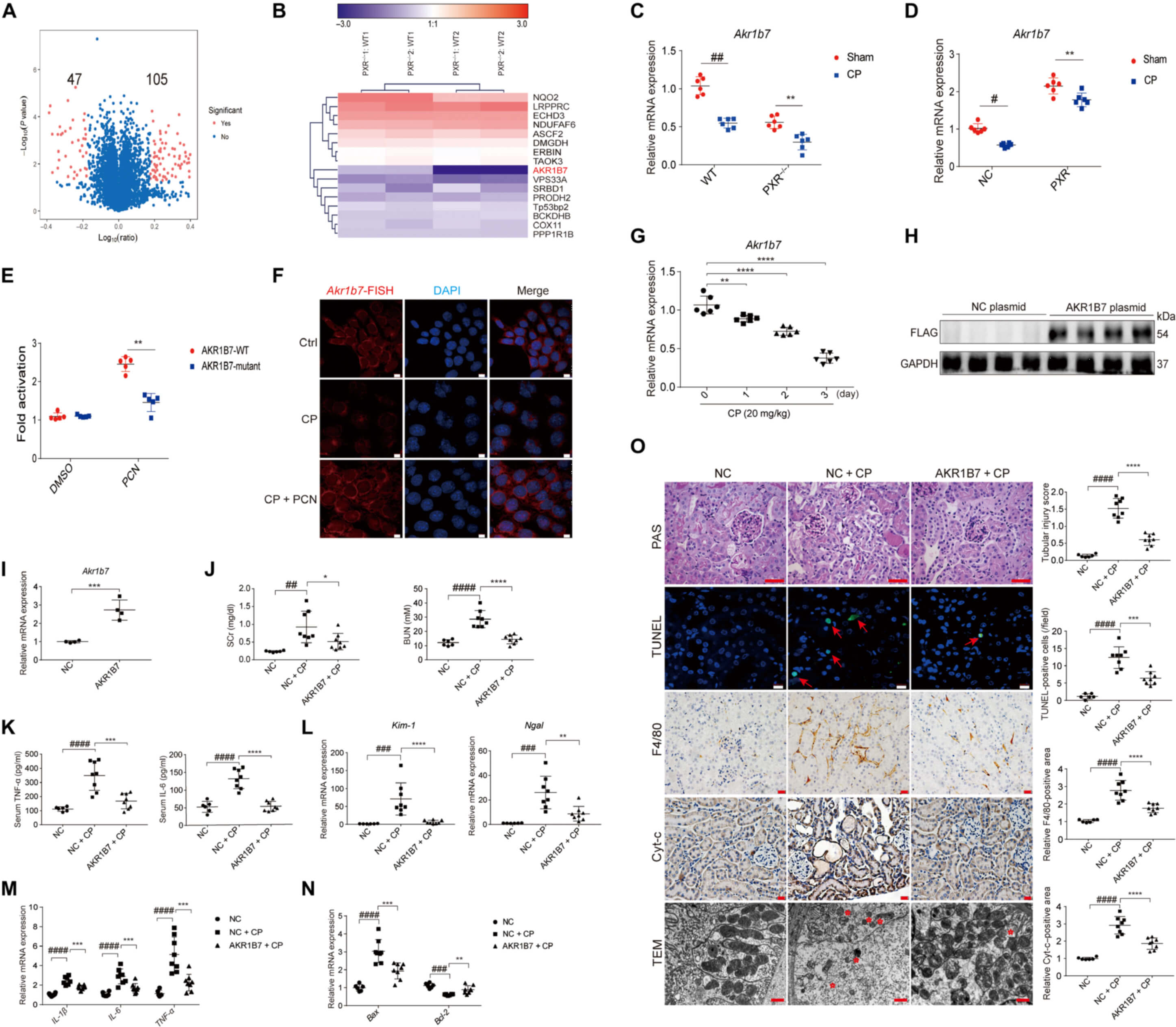
· AKR1B7 was markedly down-regulated in the kidneys of PXR−/− rats
· pGL3-Akr1b7 was activated by PCN in RTECs cotransfected with PXR vectors, whereas the mutation of Akr1b7 resulted in the loss of PXR effect.
· In situ hybridization of AKR1B7 the intensity of fluorescence decreased in cisplatin treated RTECs but recovered upon treatment with PCN
· AKR1B7 in cisplatin-induced AKI markedly improved renal function in AKI mice
· Overexpression of AKR1B7 with cisplatin treatment inhibited the release of mitochondrial Cyt-c, reduced the number of TUNEL-positive cells and F4/80-positive cells, and ameliorated
mitochondrial abnormalities
7. AKR1B7 overexpression attenuated CP-induced mitochondrial dysfunction in vitro.
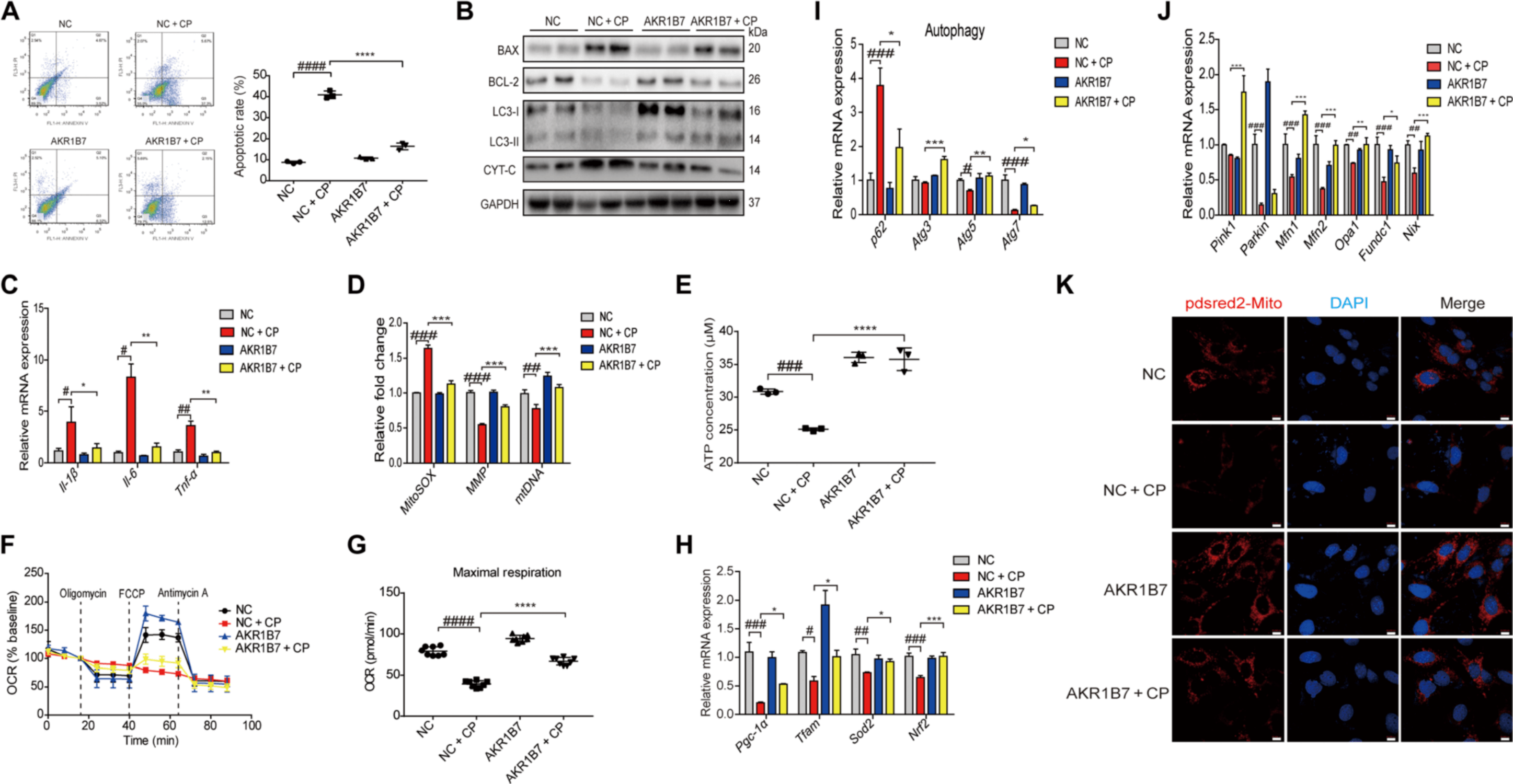
· AKR1B7 overexpression prevented cisplatin-induced cell injury—as evidenced by the decrease in cell apoptosis; the reduction of BAX, cleaved caspase-3, LC3I, LC3II, and Cyt-c; and
the restoration of BCL- 2—and
inhibited the inflammatory response
· AKR1B7 overexpression reduced cisplatin-induced accumulation of mitochondrial ROS, accompanied by a recovery of the MMP, mtDNA copy number, and ATP production in RTECs
· AKR1B7 overexpression attenuates cisplatin-induced mitochondrial dysfunction by activating mitophagy and promoting mitochondrial fusion.
8.PXR deficiency exacerbated I/R-induced AKI.
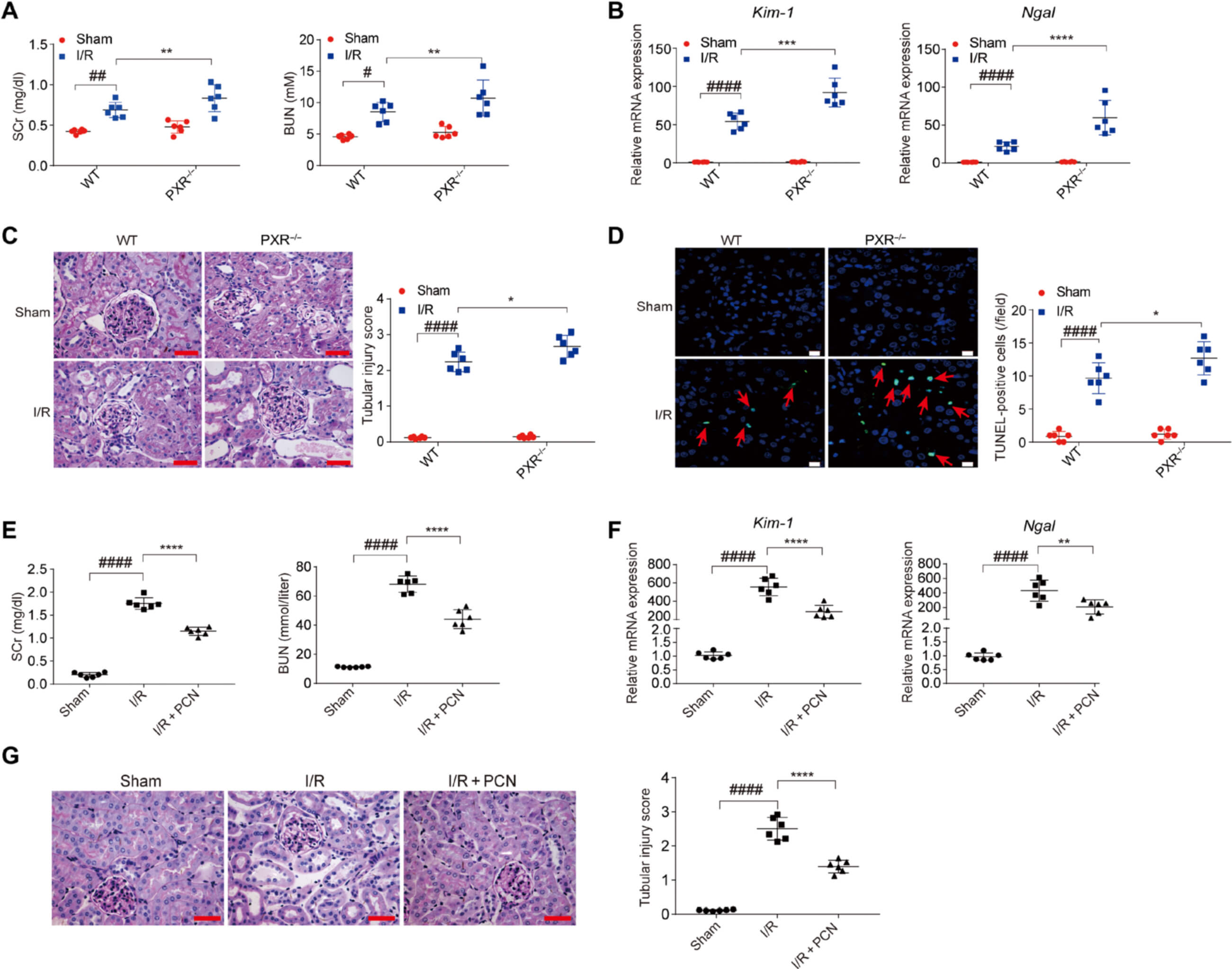
· The SCr and BUN concentrations, along with Kim-1 and Ngal mRNA expressions, were higher in the kidneys of PXR−/− rats after I/R challenge than those in the kidneys of wild-type
rats subjected to I/R in PXR
deficient (PXR−/−) rats
· PXR−/− rats exhibited exacerbated tubular injury and an increased number of TUNEL-positive cells after I/R challenge
· PXR deficiency aggravated the decrease in the expression of the mitochondrial genes in the kidneys of PXR−/− rats after I/R challenge
· The beneficial effect of PXR activation can be extended to I/R-induced AKI, suggesting a potential therapeutic use of PXR in managing AKI.
Conclusions
· This study demonstrated that the activation of the nuclear receptor PXR distinctly weakened AKI possibly by ameliorating mitochondrial dysfunction in ROS production, mitophagy, and lipid metabolism via transcriptional regulation of Akr1b7.
· It was found that PXR was robustly down-regulated and negatively correlated with renal dysfunction in human and animal kidneys with AKI. Silencing PXR in rats enhanced cisplatin-induced AKI and induced severe mitochondrial abnormalities, whereas activating PXR protected against AKI. These findings from the current research not only increase our understanding of the pathogenesis of AKI but also provide therapeutic potential by activation of the PXR/AKR1B7 pathway. Developing effective and safe agonists or activators of PXR and AKR1B7 and performing clinical trials of those agents in patients with AKI could offer a viable approach for AKI therapy.
Click Here To Check Elabscience QuicKey ELISA Kits for Renal Function

