Diabetic cardiomyopathy (DCM) represents a major complication of type 2 diabetes (T2D), with mitochondrial ros (mtROS) playing a pivotal role as both initiators and amplifiers of oxidative stress. This review examines the multifaceted contributions of mtROS to DCM pathogenesis. Upstream, mtROS production is coordinately regulated by mitochondrial bioenergetic imbalance, dynamic remodeling, and suppression of uncoupling protein 2 (UCP2) function. Downstream, mtROS propagate cellular injury through the TXNIP/NLRP3/GSDMD axis, driving inflammatory cell death, while also establishing deleterious feedback loops via epigenetic remodeling and altered secretory phenotypes. Hyperglycemia exacerbates mtROS generation through electron transport chain overload, impaired mitochondrial dynamics, compromised antioxidant defenses, and PARP-1 activation. Emerging therapeutic strategies include mitochondria-targeted antioxidants, Nrf2 activation, and mitochondrial quality control enhancers, offering promising avenues for preventing diabetes-related heart disease.
Table of Contents
1. What is diabetic heart disease?
2. Mitochondrial ros as key drivers of oxidative stress in type 2 diabetes-associated cardiomyopathy
3. How hyperglycemia exacerbates mitochondrial ros production in diabetic cardiomyocytes
4. Therapeutic strategies targeting mitochondrial ros in type 2 diabetes-related heart disease
01 What is diabetic heart disease?
Diabetes mellitus (DM) currently affects 589 million adults worldwide, representing 11.1% of the global population. This growing epidemic is projected to escalate to 853 million cases (13%) by 2050[1]. The condition imposes a substantial economic burden, accounting for nearly 10% of global health expenditure. In terms of global health impact, DM ranks as the eighth leading cause of death and the third leading contributor to years lived with disability[2]. Furthermore, diabetes significantly elevates the risk of cardiovascular type 2 diabetes complications, including ischemic heart disease, stroke, and heart failure. These cardiovascular complications are responsible for the majority of diabetes-associated morbidity and mortality.
Cardiac disease that develops as a direct consequence of diabetes mellitus in patients with type 2 diabetes (T2DM) is termed type 2 diabetes-related heart disease. This condition encompasses coronary artery disease (CAD), cardiac autonomic neuropathy (CAN), and diabetic cardiomyopathy (DCM), all of which involve molecular, structural, and functional alterations in the myocardium[3]. For example, DCM is defined by myocardial dysfunction in the absence of coronary artery disease or hypertension. Its pathogenesis involves multiple mechanisms, including hyperinsulinemia, insulin resistance, and chronic inflammation. At the cellular level, insulin resistance and type 2 diabetes-related heart disease are closely related. It disrupts lipid and glucose metabolism, promoting oxidative stress, mitochondrial dysfunction, and excessive generation of reactive oxygen species (ROS). These alterations impair cardiac function and drive pathological changes such as myocardial fibrosis, hypertrophy, and structural remodeling[4]. Advances in understanding these molecular mechanisms have translated into refined strategies for diabetes management, aimed at both alleviating complications and elucidating underlying cellular processes.
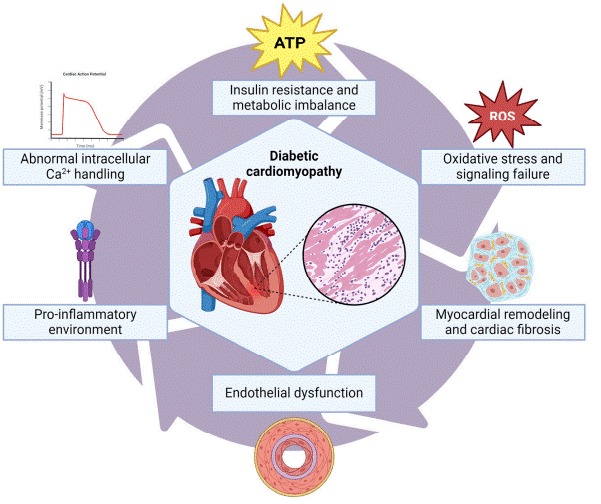
Fig. 1 Pathophysiological mechanisms in type 2 diabetes-related cardiomyopathy[4]. Chronic hyperglycemia drives oxidative stress and mtROS overproduction, activating polyol/hexosamine pathways, AGEs formation, and NF-κB inflammation. Insulin resistance increases fatty acid uptake, leading to lipotoxicity and mitochondrial dysfunction, which exacerbates mtROS generation. Elevated mtROS promote PKC dysfunction, impaired autophagy, and abnormal calcium handling, uncoupling excitation-contraction. TGF-β signaling subsequently induces myocardial remodeling and cardiac fibrosis.
02 Mitochondrial ros as key drivers of oxidative stress in type 2 diabetes-associated cardiomyopathy
As mentioned above, oxidative stress and type 2 diabetes-related heart disease are inseparable. The pathogenesis of DCM is centered on the sustained excessive production of mitochondrial ros (mtROS) induced by hyperglycemic and lipotoxic metabolic stress, whose underlying mechanism involves a multi-tiered vicious cycle of regulatory feedback. Under high glucose conditions, substrate overload in electron transport chain (ETC) complexes I and III leads to aberrant hyperpolarization of the mitochondrial membrane potential (ΔΨm). This promotes excessive reduction of the ubiquinone pool, resulting in heightened electron leakage and the generation of superoxide anions (O₂·⁻)[5]. Excessive hyperglycemia induced oxidative stress to mtDNA, resulting from its lack of histone protection and limited repair capacity, leads to defective encoding of ETC subunits. This further exacerbates respiratory chain dysfunction, thereby establishing a ROS-induced ROS release (RIRR) cycle[5]. Meanwhile, mitochondrial dynamics in the diabetic state are dysregulated, characterized by enhanced activation of the fission protein Drp1 and downregulated expression of the fusion proteins Mfn1/2 and OPA1. This imbalance drives mitochondrial fragmentation and disrupts cristae architecture, thereby exacerbating ROS leakage. Furthermore, impairment of PINK1/PARKIN-mediated selective autophagy (mitophagy) of damaged mitochondria leads to the accumulation of dysfunctional mitochondria, which serve as endogenous cellular amplifiers of sustained oxidative stress[5, 6]. Recent studies have revealed a crosstalk between methylation and oxidative stress: aberrant expression of DNA methyltransferases (DNMTs) induced by high glucose can silence antioxidant enzyme genes such as SOD2 and catalase. Conversely, mtROS can alter the methylation profile of cardiomyocytes, establishing a "metabolic memory" that propels the chronic progression of DCM[6].
At the molecular signaling level, mtROS serve not only as effectors of oxidative damage but also as second messengers that activate inflammasomes and cell death pathways. Emerging evidence has revealed that downregulation or functional inhibition of mitochondrial uncoupling protein 2 (UCP2) in the hearts of diabetic patients constitutes a critical upstream event contributing to the burst of mtROS. As an endogenous negative feedback regulator, UCP2 mildly dissipates the ΔΨm through its proton leak activity, thereby reducing the production rate of ROS from the ETC. In the context of DCM, although UCP2 expression is compensatorily upregulated, its functional capacity remains relatively insufficient. Inhibition of UCP2 exacerbates mtROS generation, which in turn activates thioredoxin-interacting protein (TXNIP), relieving its inhibition of the NLRP3 inflammasome. This cascade subsequently triggers caspase-1/GSDMD-mediated pyroptosis, ultimately exacerbating myocardial fibrosis and diastolic dysfunction[7]. Conversely, pharmacological upregulation of UCP2, such as roxadustat-mediated HIF-1α stabilization or Cirsiliol-induced activation of the PPAR-α/AMPK axis, these can restore mitochondrial membrane stability, markedly reduce mtROS production, and suppress the TXNIP/NLRP3 signaling pathway[8, 9]. Furthermore, mitochondrial oxidative stress directly reshapes the cardiac secretory phenotype. Under mtROS stress, diabetic cardiomyocytes selectively secrete inner mitochondrial membrane proteins (e.g., NDUFS and COX5A) while retaining outer membrane proteins through an unclear mechanism, constituting a non-canonical mitochondrial quality control pathway. This phenomenon occurs independently of mitophagy defects and strongly correlates with impaired cardiac function[10].
In summary, mtROS plays a dual role as both an initiator and an amplifier in DCM. Upstream, it is coordinately regulated by mitochondrial bioenergetic imbalance, dynamic remodeling, and suppressed UCP2 function. Downstream, it mediates inflammatory cell death via the TXNIP/NLRP3/GSDMD axis and forms feedback loops with epigenetic remodeling and secretory phenotype alterations.
03 How hyperglycemia exacerbates mitochondrial ros production in diabetic cardiomyocytes
3.1 Overload of the electron transport chain and superoxide generation
In the hyperglycemic state, electron overload in the mitochondrial ETC serves as a core mechanism driving ROS production in cardiomyocytes. Sustained high glucose exposure increases the supply of substrates to the tricarboxylic acid (TCA) cycle, resulting in significantly elevated levels of NADH and FADH₂. This drives a massive influx of electrons into ETC complexes I and III. When electron flow exceeds the transfer capacity of complex III, the ΔΨm becomes excessively hyperpolarized. Consequently, electrons accumulate at the coenzyme Q (CoQ) site and undergo reverse electron leak, reacting with molecular oxygen to generate superoxide anions (O₂⁻)[11]. Studies have indicated that the FMN site of complex I serves as a primary site for O₂⁻ generation, with significant contributions also arising from autoxidation of ubisemiquinone at complex III. Furthermore, high glucose-induced downregulation of mitochondrial respiratory chain complexes I and II activity exacerbates electron leakage, establishing a vicious cycle between oxidative stress and ETC dysfunction[12].
3.2 Imbalance in mitochondrial dynamics and impairment of antioxidant defense
Hyperglycemia exacerbates ROS accumulation by disrupting the balance between mitochondrial fusion and fission. High glucose conditions suppress the SIRT1/PGC-1α signaling pathway, downregulating the expression of fusion proteins Mfn2 and OPA1, while concurrently activating Drp1-mediated excessive mitochondrial fragmentation[12]. Fragmented mitochondria exhibit reduced ETC efficiency and diminished ROS clearance capacity, leading to sustained elevation of mtROS. Simultaneously, hyperglycemia depletes antioxidant reserves such as reduced GSH: on the one hand, activation of the polyol pathway consumes a large amount of NADPH, impairing the GSH regeneration capacity[5]. On the other hand, superoxide anions inhibit the activity of glucose-6-phosphate dehydrogenase (G6PD), reduce the production of NADPH, and further disrupt the antioxidant defense system. mtDNA is highly sensitive to ROS due to the lack of histone protection; oxidative damage leads to a decrease in mtDNA copy number and mutations in genes encoding respiratory chain proteins, thereby forming a positive feedback loop between ROS and mitochondrial damage[13].
3.3 PARP-1 activation and accumulation of metabolic intermediates
Hyperglycemia-induced overproduction of mtROS activates the DNA repair enzyme PARP-1, which subsequently inhibits glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity through poly(ADP-ribosyl)ation. Inactivation of GAPDH leads to the accumulation of glycolytic intermediates, including glyceraldehyde-3-phosphate (GA3P), fructose-6-phosphate (F-6-P), and glucose-6-phosphate (G-6-P), which are shunted into four pro-oxidant pathways: the polyol pathway, the hexosamine pathway, the AGEs/RAGE axis, and the PKC pathway[5]. Notably, GA3P autoxidation and AGE formation further compromise the ETC, while PKC activation upregulates NADPH oxidase (NOX) expression, synergistically amplifying oxidative stress in concert with mitochondrial ros[11]. Collectively, these metabolic derangements constitute a cascade of interconnected pathological events centered on mitochondrial ros, driving the progression of diabetic cardiomyopathy.
04 Therapeutic strategies targeting mitochondrial ros in type 2 diabetes-related heart disease
4.1 Mitochondria-targeted antioxidants
Mitochondria-targeted antioxidants (MTAs) represent a cutting-edge therapeutic strategy for the prevention and treatment of DCM by precisely scavenging ROS within mitochondria. Compounds such as MitoQ (a mitochondrial coenzyme Q10 analogue), SkQ1 (a plastoquinone derivative), and SS-31 (Elamipretide) utilize lipophilic cations (e.g., triphenylphosphonium) to penetrate the mitochondrial membrane, accumulate in the matrix, and neutralize superoxide anions. Mito-TEMPO, a mitochondria-targeted superoxide dismutase mimetic, converts superoxide anions into oxygen and hydrogen peroxide and has been demonstrated to significantly alleviate myocardial dysfunction and reduce mitochondrial ros levels in diabetic mouse models[5].
4.2 Activation of Nrf2 and induction of the endogenous antioxidant defense system
Nuclear factor erythroid 2-related factor 2 (Nrf2), as a master regulator of the cellular antioxidant response, represents a critical therapeutic target for preventing and treating diabetic cardiac injury. Upon activation, Nrf2 binds to antioxidant response elements (AREs) to upregulate the expression of antioxidant enzymes such as heme oxygenase-1 (HO-1), NAD(P)H: quinone oxidoreductase 1 (NQO1), and glutathione peroxidase 4 (GPX4), thereby enhancing mitochondrial ros scavenging capacity[14]. In diabetic cardiomyopathy, Nrf2 exhibits early adaptive upregulation; however, its expression progressively declines with disease advancement, ultimately leading to the failure of antioxidant defense mechanisms.
Natural compounds such as asiaticoside have been shown to improve mitochondrial function, enhance autophagy, and alleviate oxidative stress via the AMPK/Nrf2 pathway. Curcumin induces Nrf2 nuclear translocation and increases HO-1 expression, thereby inhibiting glucose-induced cardiomyocyte ferroptosis[14]. Furthermore, guideline-recommended agents including SGLT2 inhibitors and beta-blockers also exert cardioprotective effects, in part through Nrf2 modulation[14]. The paradigm shift from exogenous antioxidants that passively scavenge ROS toward strategies that actively activate endogenous cytoprotective pathways represents a promising new direction for the prevention and treatment of diabetic heart disease.
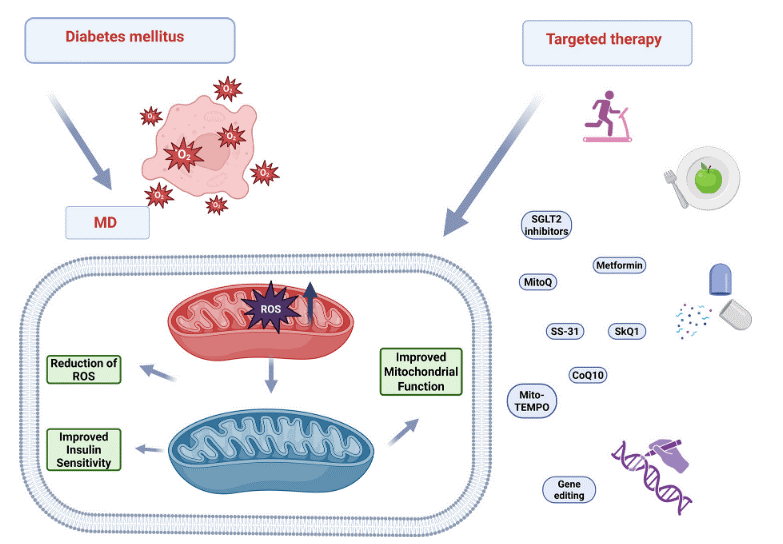
Fig. 2 Therapeutic strategies targeting mitochondrial ros in diabetic cardiomyopathy. Mitochondria-targeted antioxidants (e.g., MitoQ, CoQ10, SkQ1, SS-31, Mito-TEMPO) directly scavenge ROS within the organelle. Pharmacologic agents (metformin, SGLT2 inhibitors) and lifestyle modifications (exercise, caloric restriction) indirectly restore redox balance by enhancing mitochondrial biogenesis and insulin sensitivity. Emerging gene-editing technologies offer potential for correcting fundamental defects in mitochondrial function[5].
4.3 Mitochondrial quality control enhancers
Glucagon-like peptide-1 receptor agonists (GLP1RAs), such as exenatide and liraglutide, have demonstrated marked cardiovascular benefits in the management of T2D, with emerging evidence suggesting that these effects are closely linked to the preservation of myocardial mitochondrial quality. Large-scale cardiovascular outcome trials have confirmed that both exenatide and liraglutide significantly reduce the incidence of major adverse cardiovascular events, including the first occurrence of cardiovascular death, non-fatal myocardial infarction, or non-fatal stroke in patients with T2D[15, 16]. Subsequent mechanistic investigations have elucidated the regulatory effects of GLP1RAs on mitochondrial function. Specifically, in cardiomyocytes exposed to hyperglycemic conditions, GLP1R activation exerts antioxidant properties that attenuate mitochondrial oxidative damage[17]. Additionally, GLP1R stimulation promotes AMPK-dependent mitochondrial biogenesis and enhances fatty acid oxidation, thereby optimizing energy metabolism in cardiomyocytes[18]. Furthermore, treatment with GLP1RAs upregulates the expression of mitochondrial fusion proteins Opa1 and Mfn2, helping to preserve mitochondrial dynamics and leading to increased ATP production[19].
Furthermore, Metformin, a longstanding anti-diabetic drug, confers cardioprotective effects in diabetic cardiomyopathy, primarily through the amelioration of mitochondrial oxidative stress. Beyond its classical AMPK-dependent metabolic actions[20], metformin directly enhances mitochondrial health by restoring redox balance. In hyperglycemia-treated cardiomyocytes, metformin treatment restores the activity of mitochondrial antioxidant enzymes and suppresses the production of mitochondrial ros[21]. This improvement in oxidative status is further supported by findings that metformin normalizes mitochondrial calcium handling and prevents intracellular calcium overload in diabetic models, thereby reducing a key trigger of ROS generation and maintaining myocardial contractile function[22]. Furthermore, recent clinical evidence suggests that metformin may mitigate ROS at its source by upregulating mitophagy in mononuclear cells from T2D patients, facilitating the clearance of dysfunctional, ROS-producing mitochondria[23].
Mitochondrial ROS serve as central mediators in T2D-associated heart disease, functioning as both initiators and amplifiers of oxidative stress. Their production is governed by upstream determinants including bioenergetic dysfunction, aberrant dynamics, and impaired UCP2 activity, while downstream effectors (particularly the TXNIP/NLRP3/GSDMD inflammasome pathway) propagate inflammatory cell death and establish damaging feedback loops through epigenetic modifications. Hyperglycemia potentiates mtROS generation via electron transport chain overload, antioxidant depletion, and PARP-1 activation. Therapeutic strategies targeting mtROS, including mitochondria-targeted antioxidants, Nrf2 activators, and mitochondrial quality control enhancers, hold considerable promise. Future work should build on accurate ros detection and oxidative stress assay, with an emphasis on the spatiotemporally precise targeting of mtROS and the translation of preclinical findings into effective clinical interventions.
Elabscience® Quick Overview of Popular Products:
Table 1. Research Tools for oxidative stress assay
|
Cat. No. |
Product Name |
|
E-BC-F008 |
Mitochondrial Superoxide Fluorometric Assay Kit |
|
E-BC-K138-F |
Reactive Oxygen Species (ROS) Fluorometric Assay Kit (Green) |
|
E-BC-F005 |
Reactive Oxygen Species (ROS) Fluorometric Assay Kit (Red) |
|
E-BC-F045 |
Total Glutathione (T-GSH) And Reduced Glutathione (GSH) Fluorometric Assay Kit |
|
E-BC-K802-M |
Total Oxidant Status (TOS) Colorimetric Assay Kit |
|
E-BC-K801-M |
Total Antioxidant Status (TAS) Colorimetric Assay Kit |
|
E-BC-K025-M |
Malondialdehyde (MDA) Colorimetric Assay Kit (TBA Method) |
|
E-BC-K020-M |
Total Superoxide Dismutase (T-SOD) Activity Assay Kit (WST-1 Method) |
|
E-BC-K096-M |
Glutathione Peroxidase (GSH-Px) Activity Assay Kit |
|
E-BC-K136-M |
Total Antioxidant Capacity (T-AOC) Colorimetric Assay Kit |
|
E-BC-K035-M |
Nitric Oxide (NO) Colorimetric Assay Kit |
|
E-BC-K025-S |
Malondialdehyde (MDA) Colorimetric Assay Kit (TBA Method) |
|
E-BC-K031-M |
Catalase (CAT) Activity Assay Kit |
|
E-BC-K030-M |
Reduced Glutathione (GSH) Colorimetric Assay Kit |
|
E-BC-K074-M |
Myeloperoxidase (MPO) Activity Assay Kit |
|
E-BC-K136-S |
Total Antioxidant Capacity (T-AOC) Colorimetric Assay Kit |
|
E-BC-K097-M |
Total Glutathione (T-GSH)/Oxidized Glutathione (GSSG) Colorimetric Assay Kit |
References:
[1] MAGLIANO D J B E J, GENITSARIDI I, et al. IDF Diabetes Atlas, 11th edition [J]. INTERNATIONAL DIABETES FEDERATION, 2025.
[2] MOKDAD A H, BALLESTROS K, ECHKO M, et al. The State of US Health, 1990-2016 Burden of Diseases, Injuries, and Risk Factors Among US States [J]. JAMA-JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION, 2018, 319(14): 1444-72.
[3] RAWAL S, MANNING P, KATARE R. Cardiovascular microRNAs: as modulators and diagnostic biomarkers of diabetic heart disease [J]. CARDIOVASCULAR DIABETOLOGY, 2014, 13(1): 44.
[4] GIRALDO-GONZALEZ G C, ROMAN-GONZALEZ A, CANAS F, GARCIA A. Molecular Mechanisms of Type 2 Diabetes-Related Heart Disease and Therapeutic Insights [J]. INTERNATIONAL JOURNAL OF MOLECULAR SCIENCES, 2025, 26(10): 4548.
[5] MANOJLOVIC M, ZAFIROVIC S, NAGLIC D T, et al. Mitochondrial dysfunction, reactive oxygen species, and diabetes mellitus - A triangular relationship: A review [J]. BIOMOLECULES AND BIOMEDICINE, 2025:13145.
[6] GUO Y-Q, ZHANG J-Y, HOU P-P, et al. Cross-regulation of methylation and oxidative stress: molecular mechanisms and intervention strategies of diabetic cardiomyopathy [J]. EUROPEAN JOURNAL OF PHARMACOLOGY, 2025, 1005:178080.
[7] ZHANG L, AI C, GUO C, et al. UCP2 inhibition exaggerates diabetic cardiomyopathy by facilitating the activation of NLRP3 and pyroptosis [J]. DIABETOLOGY & METABOLIC SYNDROME, 2025, 17(1): 267.
[8] FANG T, MA C, YANG B, et al. Roxadustat improves diabetic myocardial injury by upregulating HIF-1α/UCP2 against oxidative stress [J]. CARDIOVASCULAR DIABETOLOGY, 2025, 24(1): 67.
[9] TAO J, LIU S, LING Y, et al. Cirsiliol alleviates diabetic cardiomyopathy by inhibiting oxidative stress and improving energy metabolism through the PPAR-α/AMPK pathway [J]. SCIENTIFIC REPORTS, 2025, 16(1): 2331.
[10] MIOTTO P M, BAYLISS J, FIDELITO G, et al. Diabetic heart shows preferential secretion of inner mitochondrial membrane proteins in the presence of mitochondrial oxidative stress [J]. AMERICAN JOURNAL OF PHYSIOLOGY-ENDOCRINOLOGY AND METABOLISM, 2025, 329(6): E897-E911.
[11] HUSSAIN A. Chronic hyperglycemia and cardiovascular dysfunction: an in-depth exploration of metabolic and cellular pathways in type 2 diabetes mellitus [J]. Cardiovascular diabetology Endocrinology reports, 2025, 11(1): 39.
[12] ZHI F, ZHANG Q, LIU L, et al. Novel insights into the role of mitochondria in diabetic cardiomyopathy: molecular mechanisms and potential treatments [J]. CELL STRESS & CHAPERONES, 2023, 28(6): 641-55.
[13] ZHANG Z, HUANG Q, ZHAO D, et al. The impact of oxidative stress-induced mitochondrial dysfunction on diabetic microvascular complications [J]. FRONTIERS IN ENDOCRINOLOGY, 2023, 14: 1112363.
[14] FIORI E, DAVINELLI S, FERRERA A, et al. The emerging role of Nrf2 in heart failure: From cardioprotection to therapeutic approaches [J]. ESC HEART FAILURE, 2025, 12(6): 4000-20.
[15] HOLMAN R R, BETHEL M A, MENTZ R J, et al. Effects of Once-Weekly Exenatide on Cardiovascular Outcomes in Type 2 Diabetes [J]. NEW ENGLAND JOURNAL OF MEDICINE, 2017, 377(13): 1228-39.
[16] MARSO S P, DANIELS G H, BROWN-FRANDSEN K, et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes [J]. NEW ENGLAND JOURNAL OF MEDICINE, 2016, 375(4): 311-22.
[17] ZHANG L, LI C, ZHU Q, et al. Liraglutide, a glucagon-like peptide-1 analog, inhibits high glucose-induced oxidative stress and apoptosis in neonatal rat cardiomyocytes [J]. EXPERIMENTAL AND THERAPEUTIC MEDICINE, 2019, 17(5): 3734-40.
[18] MA G, LIU Y, WANG Y, et al. Liraglutide reduces hyperglycemia-induced cardiomyocyte death through activating glucagon-like peptide 1 receptor and targeting AMPK pathway [J]. JOURNAL OF RECEPTORS AND SIGNAL TRANSDUCTION, 2020, 40(2): 133-40.
[19] NARUSE G, KANAMORI H, YOSHIDA A, et al. The intestine responds to heart failure by enhanced mitochondrial fusion through glucagon-like peptide-1 signalling [J]. CARDIOVASCULAR RESEARCH, 2019, 115(13): 1873-85.
[20] CAMERON A R, MORRISON V L, LEVIN D, et al. Anti-Inflammatory Effects of Metformin Irrespective of Diabetes Status [J]. CIRCULATION RESEARCH, 2016, 119(5): 652-65.
[21] HU M, YE P, LIAO H, et al. Metformin Protects H9C2 Cardiomyocytes from High-Glucose and Hypoxia/Reoxygenation Injury via Inhibition of Reactive Oxygen Species Generation and Inflammatory Responses: Role of AMPK and JNK [J]. JOURNAL OF DIABETES RESEARCH, 2016, 2016: 2961954.
[22] DIA M, LEON C, CHANON S, et al. Effect of Metformin on T2D-Induced MAM Ca2+ Uncoupling and Contractile Dysfunction in an Early Mouse Model of Diabetic HFpEF [J]. INTERNATIONAL JOURNAL OF MOLECULAR SCIENCES, 2022, 23(7): 3569.
[23] BHANSALI S, BHANSALI A, DUTTA P, et al. Metformin upregulates mitophagy in patients with T2DM: A randomized placebo-controlled study [J]. JOURNAL OF CELLULAR AND MOLECULAR MEDICINE, 2020, 24(5): 2832-46.

