Ferritinophagy, ferro-aging, and ferroptosis constitute a triple regulatory network governing iron-dependent cell fate. Ferritinophagy liberates redox-active Fe²⁺ from ferritin via NCOA4, fueling Fenton-driven lipid peroxidation and ferroptotic cell death. Conversely, senescent cells paradoxically accumulate massive iron but resist ferroptosis due to impaired ferritinophagy and lysosomal dysfunction, a phenomenon termed ferro-aging. Recent evidence identifies ACSL4 as a core driver of chronic ferro-aging, with vitamin C directly inhibiting ACSL4 to alleviate age-related decline in primates. Key experimental tools including ferroptosis assays, lipid peroxidation assays, GPX4 activity assays, β-galactosidase activity assays, ROS assays, and oxidative stress assays are essential for dissecting this network. Modulating these mechanisms holds therapeutic promise for cancer, neurodegeneration, and other age-related diseases.
Table of Contents
1. Ferritinophagy-mediated iron release and lipid ROS accumulation in ferroptosis
2. Quantifying labile iron pool dynamics during ferroptotic cell death
3. Crosstalk between ferroptosis and cellular senescence in aging tissues
4. GPX4 depletion and cellular vulnerability to ferroptotic stress
5. Detecting senescence-associated β-galactosidase activity in ferro-aging models
6. Ferroptosis and immunometabolic remodeling in macrophage senescence
01 Ferritinophagy-mediated iron release and lipid ROS accumulation in ferroptosis
Ferroptosis is a non-apoptotic, iron-dependent form of regulated cell death driven by catastrophic accumulation of lipid peroxides in cellular membranes[1]. Central to its initiation is ferritinophagy, the selective autophagic degradation of ferritin mediated by the cargo receptor NCOA4[2,3]. Under ferroptotic stress (e.g., erastin), NCOA4 targets ferritin heavy chain 1 (FTH1) to the autophagolysosome, where Fe³⁺ is reduced and released as redox-active Fe²⁺ into the labile iron pool (LIP)[2,3]. This Fe²⁺ drives Fenton chemistry, generating hydroxyl radicals that abstract hydrogen atoms from polyunsaturated fatty acids (PUFAs) in membrane phospholipids, leading to phospholipid hydroperoxides and chain peroxidation[1,4]. Genetic ablation of NCOA4 or autophagy blockade attenuates ferroptosis[2,3]. A comprehensive ferroptosis assay monitoring C11-BODIPY⁵⁸¹/⁵⁹¹ oxidation (for lipid ROS) and calcein-AM quenching (for LIP) simultaneously tracks ferritinophagy-dependent iron release and membrane oxidative damage[1,5].
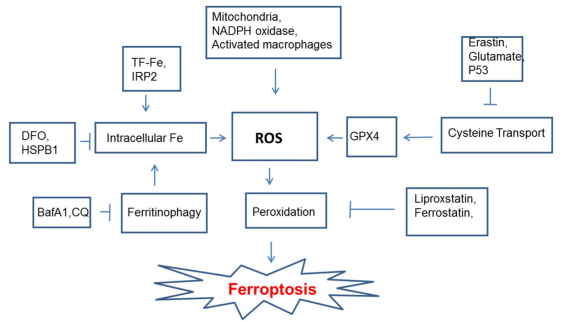
Fig. 1 Key molecular pathways regulating ferroptosis. The schematic highlights the central role of lipid peroxidation, driven by ROS from multiple sources and Fenton reactions catalyzed by intracellular iron. Iron levels are regulated by several pathways, including ferritinophagy. The antioxidant enzyme GPX4, dependent on cysteine, is a key negative regulator. Known inducers (e.g., erastin) and inhibitors (e.g., ferrostatin-1) of the process are indicated[2].
02 Quantifying labile iron pool dynamics during ferroptotic cell death
The labile iron pool (LIP) is a dynamic compartment of redox-active Fe²⁺ that acts as the immediate substrate for Fenton chemistry[3,6]. During ferroptosis, the LIP expands through two convergent mechanisms: (1) increased cellular iron import via transferrin receptor 1 (TfR1)-mediated endocytosis, and (2) NCOA4-dependent ferritinophagy, which liberates stored iron from ferritin[3].
Traditionally, the LIP has been quantified using the calcein-AM fluorescence quenching assay[3]. In recent years, RhoNox series probes, a class of Fe²⁺-specific fluorogenic dyes, have offered a more sensitive and selective alternative for real-time, live-cell imaging of labile Fe²⁺ dynamics, further underscoring the critical role of the LIP in ferroptosis initiation. Employing these methods, studies demonstrate that agents such as erastin induce a time-dependent rise in LIP that precedes detectable lipid peroxidation and cell death[3].
Although inductively coupled plasma mass spectrometry (ICP-MS) indicates that total cellular iron can increase 20- to 30-fold in senescent cells, the majority of this iron remains sequestered within ferritin. The regulatory importance of iron metabolism in ferroptosis sensitivity is further highlighted by functional evidence: overexpression of ferritin or silencing of TfR1 protects cells from ferroptosis, whereas overexpression of NCOA4 enhances cellular sensitivity to this form of cell death[3].
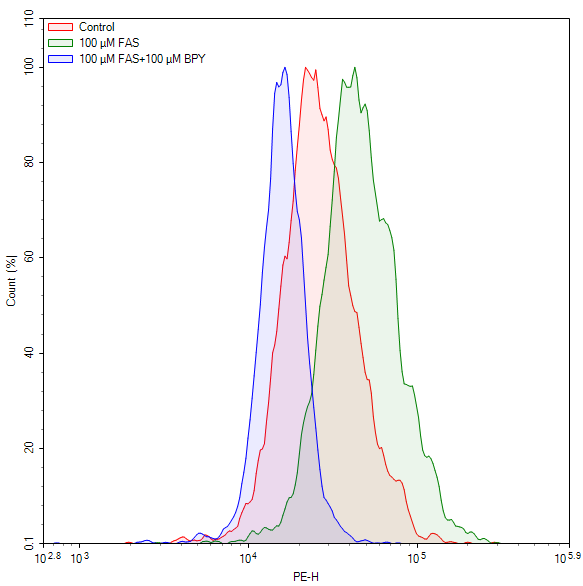
Fig. 2 Measurement of ferrous iron levels in Jurkat cells using a cell ferrous (Fe²⁺) fluorometric assay kit (E-BC-F108). Jurkat cells were treated with ferric ammonium sulfate (FAS) alone or in combination with 2,2'-bipyridyl (BPY). The results showed that BPY suppressed the FAS-induced increase in ferrous iron. (The data are provided by Elabscience)
03 Crosstalk between ferroptosis and cellular senescence in aging tissues
Cellular senescence (irreversible cell cycle arrest with SASP) and ferroptosis share a bidirectional relationship mediated by iron dyshomeostasis[3,7]. Senescent cells accumulate up to 30-fold more intracellular iron with pronounced ferritin upregulation, yet this is coupled with impaired ferritinophagy due to lysosomal dysfunction, trapping iron within ferritin[3]. This creates a perceived iron deficiency that sustains TfR1 expression and continued iron import, while conferring marked resistance to ferroptosis. This concept of “ferro-aging” (iron-driven senescence-associated dysfunction) positions ferroptosis modulation as a therapeutic strategy for age-related diseases[7,8]. Beyond the acute GPX4-dependent ferroptosis pathway, a distinct chronic ferro-aging program driven by ACSL4 has been identified ACSL4, the rate-limiting enzyme for PUFA esterification into membrane phospholipids, progressively increases with age and generates substrate for sustained low-level lipid peroxidation[9]. Notably, vitamin C directly binds and inhibits ACSL4 at residues Thr278/Ser279/Thr469, also activating NRF2[9]. In a 40-month primate study, long-term vitamin C supplementation attenuated ferro-aging features across multiple tissues, reduced biological age (multi-omics clocks), preserved brain structure, and improved metabolic parameters, offering a safe, translatable intervention[9].
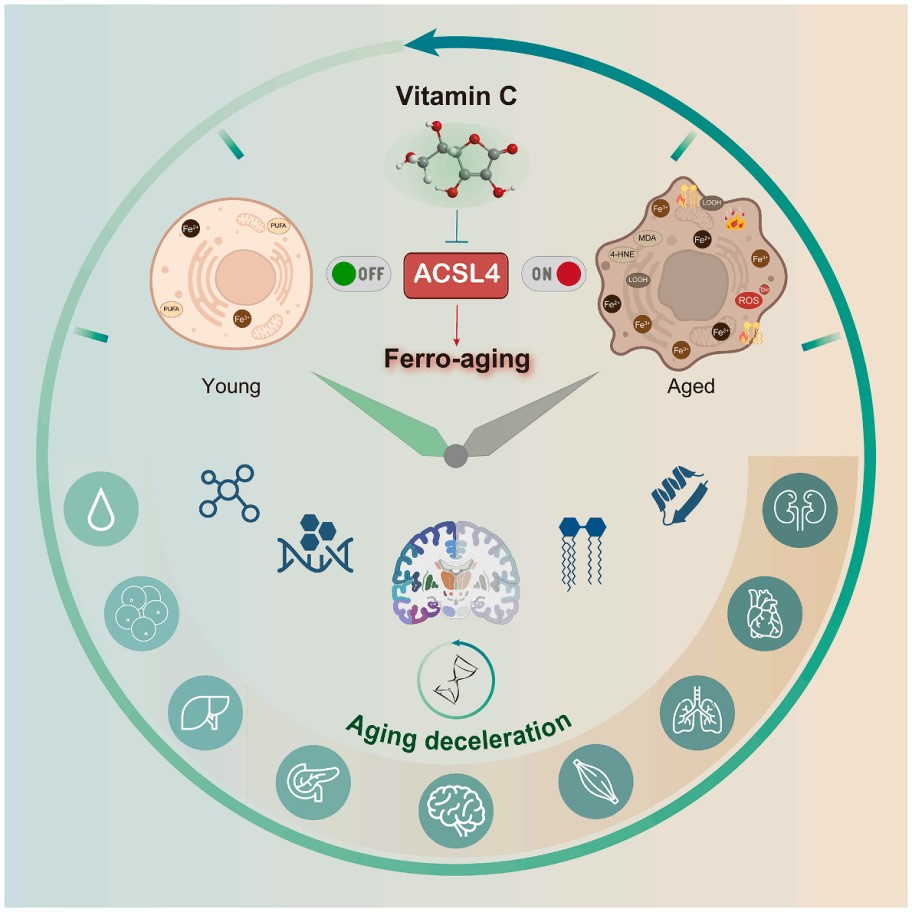
Fig. 3 The mechanism of Vitamin C in inhibiting ACSL4 to mitigate ferro-aging in primates. The diagram utilizes a clock face metaphor to contrast "Young" and "Aged" physiological states, emphasizing that the intervention (targeting the ACSL4 pathway) promotes "Aging deceleration." Various icons representing cellular structures, molecules, and physiological processes are arranged around the periphery to symbolize the comprehensive impact of aging and its deceleration[9].
04 GPX4 depletion and cellular vulnerability to ferroptotic stress
Glutathione peroxidase 4 (GPX4) is the master ferroptosis suppressor, uniquely reducing phospholipid hydroperoxides to non-toxic lipid alcohols using GSH as a cofactor[4]. Its catalytic selenocysteine is alkylated by RSL3, causing immediate inactivation[4]. The GPX4 activity assay measures NADPH consumption during reduction of phosphatidylcholine hydroperoxides in a coupled spectrophotometric system[4]. GPX4 depletion occurs via multiple routes: class I inducers (e.g., erastin) deplete GSH by inhibiting system xc⁻; class II inducers (e.g., RSL3) modify the active site; class III inducers (e.g., FIN56) promote GPX4 degradation[1,4]. However, GPX4 inactivation alone does not guarantee ferroptosis; parallel surveillance systems (FSP1/CoQ10, GCH1/BH4) generate lipophilic radical-trapping antioxidants[1]. Therefore, an integrated oxidative stress assay measuring the GSH/GSSG ratio alongside GPX4 activity provides a more complete assessment of cellular ferroptosis vulnerability[4].
05 Detecting senescence-associated β-galactosidase activity in ferro-aging models
The senescence-associated β-galactosidase (SA-β-gal) assay using X-Gal at pH 6.0 remains the standard biomarker for identifying senescent cells in culture and tissue sections, relying on the expanded lysosomal compartment[3]. In ferro-aging studies, SA-β-gal staining spatially maps senescent populations under iron dysregulation: in aged mouse liver, ~10% of cells are SA-β-gal-positive, matching ferritin-enriched cells and linking senescence to iron overload in vivo. SA-β-gal positivity exceeds 80% by day 10 post-irradiation in mouse embryonic fibroblasts[3]. Caution is needed for immune cells (e.g., macrophages), as activation can elevate lysosomal β-galactosidase independently of senescence. For rigorous identification, complement SA-β-gal staining with p16^INK4a, p21^WAF1/CIP1, TIF, and SASP profiling.
Alternatively, flow cytometry using fluorogenic substrates at pH 6.0 enables rapid, quantitative detection of β-galactosidase activity in single cells, facilitating high-throughput senescence assessment in ferro-aging models. This method is compatible with surface marker co-staining to exclude non-senescent lysosomal activation in immune subsets.
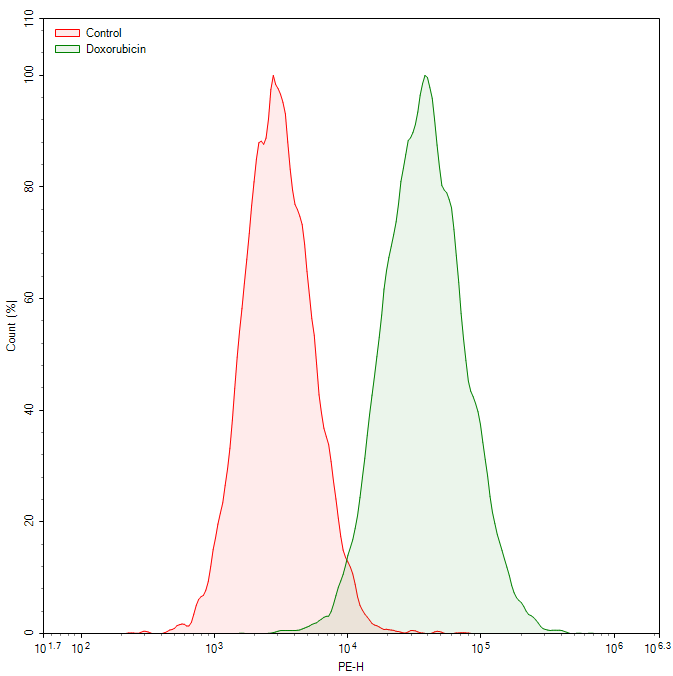
Fig. 4 Flow cytometric analysis of doxorubicin-induced senescence in WI-38 cells. SA-β-galactosidase (SA-β-gal) activity was detected using a cellular senescence fluorometric assay kit (E-BC-F086). Compared to the untreated control group, the doxorubicin-treated group shows a significant increase in SA-β-gal activity, as indicated by the marked rightward shift of the fluorescence peak. (The data are provided by Elabscience)
06 Ferroptosis and immunometabolic remodeling in macrophage senescence
Macrophage senescence features durable cell cycle arrest, pro-inflammatory SASP, impaired phagocytosis, altered polarization, and metabolic reprogramming[7]. The intersection of ferroptosis with macrophage senescence is a critical nexus for age-related immunometabolic dysfunction. Aged macrophages exhibit mitochondrial dysfunction, reduced NAD⁺ synthesis, and compromised antioxidant defenses, which enhance oxidative stress vulnerability[7]. Age-dependent intracellular iron accumulation in macrophages (driven by increased TfR1 and impaired ferritinophagy) creates conditions permissive for ferroptosis[3]. Senescent macrophage polarization shifts toward M1 or M2 depending on the microenvironment, linking iron handling and lipid metabolism[7]. In atherosclerosis, oxidized LDL-induced macrophage senescence elevates SA-β-gal activity and heightens ferroptosis sensitivity, contributing to plaque instability[7]. In KRAS-driven lung cancer, senescent alveolar macrophages accumulate in premalignant lesions and exhibit ferroptosis resistance via GPX4 and SLC7A11 upregulation, promoting an immunosuppressive tumor microenvironment[8]. The ROS assay using DCFH-DA ,combined with the lipid peroxidation assay using C11-BODIPY⁵⁸¹/⁵⁹¹, are essential for characterizing redox status in senescent macrophages[4]. Ferroptosis-based senolysis represents a promising therapeutic frontier for age-related diseases[8].
Conclusion
Ferritinophagy supplies Fe²⁺ for Fenton-driven lipid peroxidation. Ferroptosis is acute iron-dependent cell death, while ferro-aging is a chronic senescent state with iron overload but ferroptosis resistance. ACSL4 drives ferro-aging, and vitamin C directly inhibits ACSL4[9]. Accurate assessment requires ferroptosis assays, lipid peroxidation assays, GPX4 activity assays, β-galactosidase activity assays, ROS/reactive oxygen species assays, and oxidative stress assays. Inducing ferroptosis in cancer or inhibiting ferroptosis/ferro-aging in degenerative diseases represents opposite sides of the same iron-based therapeutic coin.
Table 1. Comparison of Ferritinophagy, Ferroptosis, and Ferro-aging
|
Process |
Mechanism |
Detection |
Key Feature |
|
Ferritinophagy |
NCOA4-mediated autophagic degradation of ferritin → releases Fe²⁺ into LIP |
NCOA4/LC3 colocalization; ferritin degradation (WB); autophagy flux assays |
Iron supplier; triggers both ferroptosis and ferro-aging |
|
Ferroptosis |
Fe²⁺ drives Fenton reaction → PUFA peroxidation → membrane rupture; GPX4 inactivation |
Ferroptosis assay(C11-BODIPY + calcein-AM); lipid peroxidation assay(MDA/4-HNE); GPX4 activity assay; ROS assay; GSH/GSSG) |
Acute cell death; requires ferritinophagy-derived iron |
|
Ferro-aging |
Chronic iron overload + ACSL4 upregulation → sustained low-grade lipid peroxidation → lysosomal dysfunction → senescence |
β-galactosidase activity(SA-β-gal); iron accumulation; ACSL4 activity; SASP factors |
Chronic senescent state; paradoxically ferroptosis-resistant |
Quick Overview of Popular Products:
Table 2. Reagents for Ferritinophagy, Ferroptosis, and Ferro-aging
|
Cat. No. |
Product Name |
|
E-BC-F003 |
Lipid Peroxide (LPO) Fluorometric Assay Kit |
|
E-BC-K025-M |
Malondialdehyde (MDA) Colorimetric Assay Kit (TBA Method) |
|
E-BC-K814-M |
Enhanced Cell Malondialdehyde (MDA) Colorimetric Assay Kit |
|
E-EL-0128 |
4-HNE(4-Hydroxynonenal) ELISA Kit |
|
E-BC-F108 |
Cell Ferrous (Fe2+) Fluorometric Assay Kit |
|
E-BC-K881-M |
Cell Ferrous Iron Colorimetric Assay Kit |
|
E-BC-K138-F |
Reactive Oxygen Species (ROS) Fluorometric Assay Kit (Green) |
|
E-BC-F005 |
Reactive Oxygen Species (ROS) Fluorometric Assay Kit (Red) |
|
E-BC-F066 |
Cystine Uptake Fluorometric Assay Kit |
|
E-BC-F045 |
Total Glutathione (T-GSH) And Reduced Glutathione (GSH) Assay Kit |
|
E-BC-F086 |
Cellular Senescence Fluorometric Assay Kit |
|
E-BC-K883-M |
Glutathione Peroxidase 4 (GPX4) Activity Assay Kit |
|
E-BC-K771-M |
Lactate Dehydrogenase (LDH) Cytotoxicity Colorimetric Assay Kit |
|
E-CK-A362 |
Enhanced Cell Counting Kit 8 (WST-8/CCK8) |
References:
[1] Dixon SJ, Olzmann JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. 2024;25(6):424-442. DOI: 10.1038/s41580-024-00703-5
[2] Latunde-Dada GO. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gen Subj. 2017;1861(8):1893-1900. DOI: 10.1016/j.bbagen.2017.05.019
[3] Masaldan S, Clatworthy SAS, Gamell C, Smith MA, Bush AI, Lesniak W, et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018;14:100-115. DOI: 10.1016/j.redox.2017.08.015
[4] Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic Biol Med. 2020;152:175-185. DOI: 10.1016/j.freeradbiomed.2020.02.027
[5] Zhou RP, Chen Y, Wei X, Yu B, Xiong ZG, Lu C, et al. Novel insights into ferroptosis: Implications for age-related diseases. Theranostics. 2020;10(26):11976-11997. DOI: 10.7150/thno.50663
[6] Mazhar M, Ud Din A, Ali H, Yang G, Ren W, Wang L, et al. Implication of ferroptosis in aging. Cell Death Discov. 2021;7:149. DOI: 10.1038/s41420-021-00553-6
[7] Wang LL, Hong WX, Zhu H, He QJ, Yang B, Wang JJ, et al. Macrophage senescence in health and diseases. Acta Pharm Sin B. 2024;14(4):1508-1524. DOI: 10.1016/j.apsb.2024.01.008
[8] Liang D, Minikes AM, Jiang X. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell. 2023;186(13):2748-2764.e22. DOI: 10.1016/j.cell.2023.05.003
[9] Liu G, Qu J, Zhang W, et al. Vitamin C inhibits ACSL4 to alleviate ferro-aging in primates. Cell Metab. 2026;38(4):635-664. DOI: 10.1016/j.cmet.2026.02.001

