Ischemia, as exemplified by myocardial infarction, creates a dynamic microenvironment wherein macrophage functional polarization is intrinsically governed by a core metabolic axis balancing glycolysis and fatty acid oxidation. Following ischemic injury, infiltrating macrophages undergo temporally regulated metabolic reprogramming. The early pro-inflammatory state is fueled by aerobic glycolysis, driven by hypoxia-inducible factor (HIF)-1α and supporting host defense and debris clearance. The essential transition to a reparative phenotype requires a metabolic switch toward fatty acid oxidation and oxidative phosphorylation, orchestrated by AMPK and SIRT1. This critical glucose-lipid metabolic switch is centrally controlled by the antagonistic AMPK-mTORC1 signaling axis and their downstream metabolic enzymes. Conversely, persistent glycolytic metabolism sustains a pro-inflammatory state, thereby driving adverse cardiac remodeling.
In this context, we highlight macrophage metabolic reprogramming as a pivotal determinant of post-ischemic outcome and a promising therapeutic frontier. Targeting this glycolipid interplay, for example by inhibiting mTORC1 and glycolysis or by enhancing AMPK, SIRT1, and fatty acid oxidation pathways, represents a novel strategy for myocardial ischemia treatment. This approach aims to promote the reparative macrophage phenotype, resolve maladaptive inflammation, and improve cardiac repair.
Table of Contents
1. Glycolysis and fatty acid oxidation in ischemic macrophage polarization
2. Metabolic reprogramming during macrophage polarization in ischemia
3. Enzymatic control of glycolipid metabolism in ischemic macrophages
4. Hypoxia-driven metabolic regulation of macrophage polarization
5. AMPK-mTOR crosstalk in glycolipid metabolic reprogramming
6. SIRT1 modulation of glycolysis–lipid metabolism interplay under ischemia
01 Glycolysis and fatty acid oxidation in ischemic macrophage polarization
Glycolysis and fatty acid oxidation (FAO) form a core metabolic axis that regulates macrophage polarization into pro-inflammatory (M1) and reparative (M2) phenotypes following ischemic injury, such as myocardial infarction[1]. The initial pro-inflammatory M1 polarization is characterized by a pronounced transition to aerobic glycolysis (the Warburg effect), enabling rapid ATP production and biosynthesis. Disruption of the tricarboxylic acid (TCA) cycle leads to accumulation of metabolites, including citrate and succinate, which stabilize the M1 phenotype and enhance the generation of nitric oxide, reactive oxygen species, and pro-inflammatory cytokines like IL-1β, thereby driving tissue debridement[2]. During the resolution phase, metabolic reprogramming towards FAO and oxidative phosphorylation (OXPHOS) underpins the reparative M2 phenotype, a shift that can be quantified using fatty acid oxidation assay (FAO assay) and oxidative phosphorylation assay. Signaling via the IL-4 and IL-13-activated STAT6 axis upregulates FAO machinery and promotes mitochondrial biogenesis, supplying sustained energy and facilitating the expression of anti-inflammatory and pro-reparative genes[3]. Critically, this metabolic shift is instructive, as inhibiting FAO, a finding robustly demonstrated by FAO assay, impedes M2 polarization. Thus, the temporal switch from glycolysis to FAO serves as a fundamental driver of the macrophage phenotypic transition from inflammation to repair in ischemia.
02 Metabolic reprogramming during macrophage polarization in ischemia
Metabolic reprogramming is a fundamental mechanism underlying macrophage plasticity, driving the phenotypic switch between pro-inflammatory (M1) and reparative (M2) states within the ischemic microenvironment[4]. Following injury, infiltrating macrophages undergo a tightly regulated metabolic shift. The initial pro-inflammatory M1 response is fueled by enhanced glycolysis, a fragmented TCA cycle, and an activated pentose phosphate pathway (PPP). This state is characterized by metabolite accumulation, including succinate, which stabilizes hypoxia-inducible factor-1α (HIF-1α) to promote a pro-inflammatory gene program, and increased NADPH production, supporting reactive oxygen species (ROS) generation[5]. In contrast, the transition to a pro-reparative M2 phenotype is coupled to a metabolic reprogramming towards FAO and enhanced oxidative phosphorylation (OXPHOS), a functional shift that can be directly measured by oxidative phosphorylation assay (OXPHOS assay). M2 macrophages utilize an intact TCA cycle and efficient mitochondrial respiration, as evidenced by OXPHOS assay, to support sustained anti-inflammatory and tissue-remodeling functionss[5, 6]. This metabolic switch, further reinforced by processes such as efferocytosis, positions macrophage metabolic flexibility as a promising therapeutic target for improving repair in ischemic diseases[7].
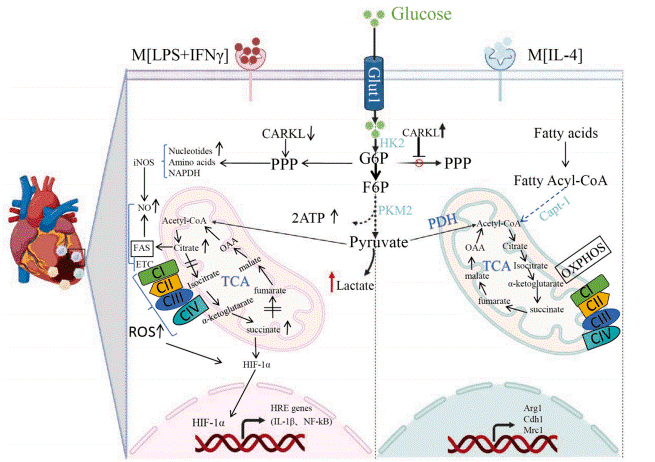
Fig. 1 Macrophage metabolic reprogramming and inflammatory features[5]. A schematic diagram comparing the metabolic profiles of classically activated (M1) and alternatively activated M2 macrophages, highlighting distinct glucose and fatty acid metabolism pathways that contribute to their respective inflammatory and anti-inflammatory phenotypes.
03 Enzymatic control of glycolipid metabolism in ischemic macrophages
The phenotypic fate of macrophages in the ischemic niche is molecularly steered by key metabolic enzymes that act as decisive switches between glycolytic and oxidative programs. Central to the pro-inflammatory M1 metabolism is the glycolytic enzyme pyruvate kinase M2 (PKM2). Its dimeric form, predominant in M1 cells, favors aerobic glycolysis and can translocate to the nucleus to cooperate with HIF-1α, augmenting inflammatory gene expression[5]. Concurrently, pyruvate dehydrogenase kinase 1 (PDK1), induced by HIF-1α, phosphorylates and inactivates the pyruvate dehydrogenase complex[8]. This critical shunt diverts pyruvate from mitochondrial oxidation to lactate production, cementing the glycolytic state. Conversely, the reparative M2 phenotype is characterized by enhanced FAO, governed by carnitine palmitoyltransferase 1 (CPT1). CPT1 activity, facilitating mitochondrial fatty acyl-CoA import, is essential for alternative activation. Its function is potently regulated by acetyl-CoA carboxylase (ACC), which synthesizes malonyl-CoA, a physiologic inhibitor of CPT1[9]. Thus, the balance between ACC-mediated inhibition and AMPK-driven relief of this inhibition fine-tunes FAO flux. Furthermore, enzymes like succinate dehydrogenase (SDH) in the TCA cycle create feedback loops; its repression in M1 macrophages leads to succinate accumulation, which stabilizes HIF-1α and further propels glycolysis[2]. This interconnected enzymatic network, integrating hypoxic and inflammatory signals, offers precise targets for metabolically driving macrophage polarization towards a tissue-reparative state in ischemia.
%20activities%20during%20macrophage%20M1%20polarization_.png)
Fig. 2 Metabolic enzyme and fatty acid oxidation (FAO) activities during macrophage M1 polarization. (A) Pyruvate kinase (PK) activity in M0 and M1 RAW264.7 macrophages. (B) FAO activity in M0 and M1 THP-1 macrophages (The datas are provided by Elabscience).
04 Hypoxia-driven metabolic regulation of macrophage polarization
Hypoxia, as a key characteristic of ischemic microenvironment, serves as a defining and instructive signal within the ischemic niche, driving metabolic reprogramming to dictate macrophage functional polarization[10]. This regulation is principally orchestrated by the hypoxia-inducible factor (HIF) family of transcription factors, which are stabilized upon oxygen deprivation. HIF-1α is a pivotal mediator of the pro-inflammatory M1 phenotype. It transcriptionally activates a suite of glycolytic genes, shunting metabolism towards aerobic glycolysis to meet the biosynthetic and energetic demands of inflammation[11, 12]. Concurrently, HIF-1α suppresses oxidative metabolism, including FAO, thereby cementing the glycolytic M1 state. Notably, HIF-1α stabilization and its metabolic program can be triggered not only by hypoxia but also by inflammatory stimuli like LPS via mitochondrial ROS, indicating a convergence of hypoxic and damage signals in ischemic tissue[13].
In contrast, HIF-2α often exhibits a divergent role, promoting an anti-inflammatory, reparative M2 polarization[14]. It enhances oxidative metabolism by upregulating key FAO enzymes, supporting mitochondrial oxidative phosphorylation (OXPHOS) and providing sustained energy for tissue remodeling functions[12]. The temporal and contextual balance between HIF-1α and HIF-2α activity is therefore critical for determining the macrophage phenotypic trajectory.
Furthermore, the hypoxic microenvironment generates metabolic signals that feed back onto macrophages. Notably, lactate, which accumulates as a consequence of glycolytic metabolism, acts as a paracrine signaling molecule. It can be sensed via the G-protein coupled receptor GPR81 on macrophages, promoting an M2-like phenotype and supporting repair processes[15, 16]. Thus, hypoxia-driven metabolic regulation operates through both cell-intrinsic transcriptional programs, mediated by HIF isoforms, and extrinsic signals from the metabolic milieu, collectively fine-tuning macrophage polarization in ischemic disease. Targeting these specific hypoxic signaling hubs offers a promising strategy for therapeutically modulating macrophage function.
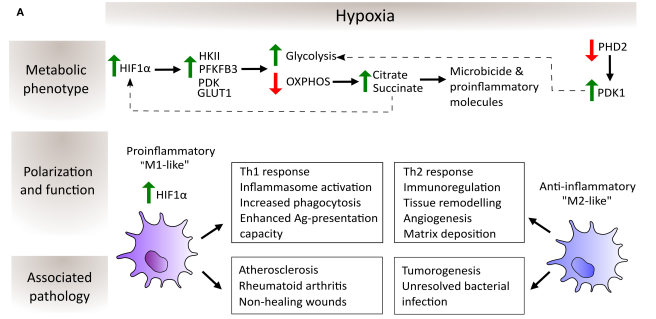
Fig. 3 Schematic depiction of the regulation of macrophage metabolic phenotype, polarization and function by hypoxia[10]. Hypoxia stabilizes HIF-1α, reprogramming macrophage metabolism from OXPHOS to glycolysis. This metabolic shift, mediated by enzymes like PDK1, drives divergent polarization into pro-inflammatory (M1-like) or anti-inflammatory (M2-like) phenotypes, contributing to diseases such as atherosclerosis and cancer.
05 AMPK-mTOR crosstalk in glycolipid metabolic reprogramming
The AMPK-mTOR signaling axis serves as a central metabolic switch governing glycolipid metabolic reprogramming in macrophages, a process critical under conditions like ischemic stress[17, 18]. This integrated signaling network, centered on the antagonism between AMP-activated protein kinase (AMPK) and the mechanistic target of rapamycin complex 1 (mTORC1), dictates the cellular energy balance and functional phenotype[19]. As a cellular energy sensor, AMPK activation during low ATP states promotes catabolic pathways, including FAO and OXPHOS, metabolically underpinning an anti-inflammatory, reparative (M2) macrophage state. Conversely, the mTORC1 limb of this axis transduces nutrient and growth signals to drive anabolic metabolism, such as glycolysis and lipogenesis, thereby supporting the energetic basis of the pro-inflammatory (M1) phenotype[20].
The crosstalk within the AMPK mTOR signaling pathway is direct and reciprocal, forming a precise regulatory circuit. AMPK activity phosphorylates and inhibits key nodes like the mTORC1 subunit Raptor while activating the mTORC1 suppressor TSC2[21, 22]. This suppression of mTORC1 by AMPK concurrently dampens glycolytic flux and relieves its repression on FAO, steering the integrated signaling output toward an oxidative, M2-favoring program. In contrast, dominant mTORC1 signaling reinforces glycolysis and can inhibit AMPK, creating a positive feedback loop that stabilizes the M1 state. Thus, the dynamic equilibrium of the AMPK-mTOR axis ensures macrophage metabolism and function are matched to the tissue's energetic and inflammatory status. Disruption of this integrated signaling, as seen when AMPK-mediated inhibition of mTORC1 is impaired, leads to aberrant metabolic and immune responses[23]. Consequently, pharmacological modulation of the AMPK-mTOR signaling axis represents a promising therapeutic strategy to guide macrophage polarization in ischemic diseases.
06 SIRT1 modulation of glycolysis–lipid metabolism interplay under ischemia
Sirtuin 1 (SIRT1) functions as a pivotal metabolic coordinator, integrating NAD+-dependent signaling to direct the interplay between glycolysis and lipid metabolism. This interplay underpins macrophage fate determination in ischemic tissues[24]. Governed by cellular NAD+ availability, SIRT1 acts as a central sensor that translates the metabolic state of the ischemic milieu into specific epigenetic and transcriptional programs. A primary mechanism involves the differential deacetylation of key transcriptional regulators. SIRT1 deacetylates and activates peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), a master driver of mitochondrial biogenesis and fatty acid oxidation (FAO), thereby promoting the oxidative metabolic program essential for the reparative M2 phenotype[25]. Conversely, it targets HIF-1α for deacetylation, promoting its degradation and the subsequent suppression of glycolytic gene expression, which helps resolve pro-inflammatory M1 signaling[26].
This regulatory capacity is amplified through a synergistic positive feedback loop with AMP-activated protein kinase (AMPK). AMPK activation elevates NAD+ levels, thereby stimulating SIRT1 activity. In turn, SIRT1 deacetylates and activates LKB1, the upstream kinase responsible for AMPK phosphorylation[27]. This reciprocal reinforcement solidifies a metabolic shift towards FAO and oxidative phosphorylation while concurrently inhibiting glycolytic flux. Within the ischemic context, the integrity of this SIRT1-governed network is critical for effective phenotypic transition. For instance, impaired NAD+ generation during efferocytosis disrupts SIRT1 activation, leading to defective anti-inflammatory reprogramming and worsened post-ischemic outcomes[28].
Thus, SIRT1 operates as a critical rheostat, fine-tuning the metabolic switch from glycolysis to lipid oxidation to guide macrophages from a pro-inflammatory to a tissue-reparative state. This highlights its therapeutic potential in ischemic pathologies, which can be further investigated and validated through targeted SIRT1 assay[29].
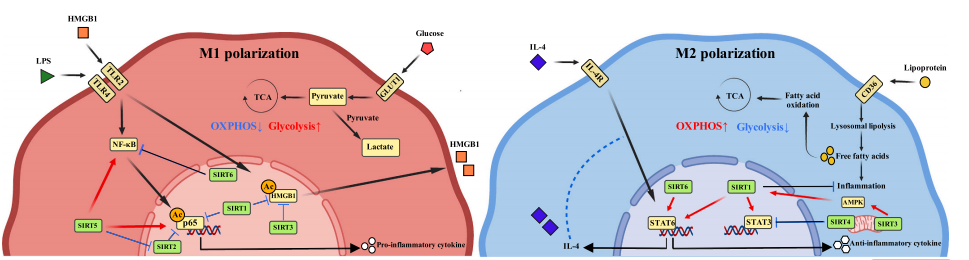
Fig. 4 Regulatory pathways of the Sirtuin family in macrophage polarization[24]. Comparative schematic of macrophage polarization: Inflammatory M1 (left, red) activated by LPS/IFN-γ relies on HIF-1α-driven glycolysis. Anti-inflammatory M2 (right, blue) activated by IL-4 utilizes PPARδ/STAT6-mediated fatty acid oxidation and OXPHOS.
Quick Overview of Popular Products:
Table 1. Reagents for Macrophage Metabolic Reprogramming and Ischemic Disease Research
|
Cat. No. |
Product Name |
|
E-BC-K784-M |
Fatty Acid Oxidation (FAO) Colorimetric Assay Kit |
|
E-BC-F078 |
Mitochondrial Stress Fluorometric Assay Kit |
|
E-BC-F056 |
Sirtuin 1 (SIRT-1) Activity Assay Kit |
|
E-BC-F201 |
Enhanced ATP Chemiluminescence Assay Kit |
|
E-EL-H6066 |
Human HIF-1α (Hypoxia Inducible Factor 1 Alpha) ELISA Kit |
|
E-EL-R0513 |
Rat HIF-1α (Hypoxia Inducible Factor 1 Alpha) ELISA Kit |
|
E-EL-M0687 |
Mouse HIF-1α (Hypoxia Inducible Factor 1 Alpha) ELISA Kit |
|
E-BC-K611-M |
Pyruvate Kinase (PK) Activity Assay Kit |
|
E-BC-F070 |
Enhanced Oxygen Consumption Rate (OCR) Fluorometric Assay Kit |
|
XJM004 |
RAW 264.7 Polarized M1 Macrophage Induction and Identification Kit |
|
E-EL-M0043 |
Mouse IL-4 (Interleukin 4) ELISA Kit |
|
E-EL-H0101 |
Human IL-4 (Interleukin 4) ELISA Kit |
|
E-BC-K649-M |
Succinate Dehydrogenase (SDH) Activity Assay Kit |
|
E-BC-K044-M |
L-Lactic Acid (LA) Colorimetric Assay Kit |
References:
[1] Batista-Gonzalez, A., et al., New Insights on the Role of Lipid Metabolism in the Metabolic Reprogramming of Macrophages. Frontiers in Immunology, 2020. 10: p. 2993.
[2] Thorp, E.B., Macrophage Metabolic Signaling during Ischemic Injury and Cardiac Repair. Immunometabolism, 2021. 3(2): p. e210013.
[3] Zhang, S., et al., Immunometabolism of Phagocytes and Relationships to Cardiac Repair. Frontiers in Cardiovascular Medicine, 2019. 6: p. 42.
[4] Ji, Y., et al., Macrophage Polarization: Molecular Mechanisms, Disease Implications, and Targeted Therapeutic Strategies. Frontiers in Immunology, 2025. 16: p. 1732718.
[5] Ao-Di, F., et al., Advances in Macrophage Metabolic Reprogramming in Myocardial Ischemia-Reperfusion. Cellular Signalling, 2024. 123: p. 111370.
[6] Wong, A., et al., Macrophage Energy Metabolism in Cardiometabolic Disease. Molecular and Cellular Biochemistry, 2025. 480(3): p. 1763-1783.
[7] Zhang, S., et al., Targeting NPM1 Epigenetically Promotes Postinfarction Cardiac Repair by Reprogramming Reparative Macrophage Metabolism. Circulation, 2024. 149(25): p. 1982-2001.
[8] Semba, H., et al., HIF-1α-PDK1 Axis-Induced Active Glycolysis Plays an Essential Role in Macrophage Migratory Capacity. Nature Communications, 2016. 7: p. 11635.
[9] Nomura, M., et al., Fatty Acid Oxidation in Macrophage Polarization. Nature Immunology, 2016. 17(3): p. 216-217.
[10] Díaz-Bulnes, P., et al., Crosstalk Between Hypoxia and ER Stress Response: A Key Regulator of Macrophage Polarization. Frontiers in Immunology, 2020. 10: p. 2951.
[11] Qiu, B., et al., Hypoxia Inducible Factor-1α Is an Important Regulator of Macrophage Biology. Heliyon, 2023. 9(6): p. e17167.
[12] Zuo, W., et al., Macrophage-Driven Cardiac Inflammation and Healing: Insights from Homeostasis and Myocardial Infarction. Cellular & Molecular Biology Letters, 2023. 28(1): p. 81.
[13] Fuhrmann, D.C., I. Wittig, and B. Brüne, TMEM126B Deficiency Reduces Mitochondrial SDH Oxidation by LPS, Attenuating HIF-1α Stabilization and IL-1β Expression. Redox Biology, 2019. 20: p. 204-216.
[14] DeBerge, M., et al., Hypoxia-Inducible Factors Individually Facilitate Inflammatory Myeloid Metabolism and Inefficient Cardiac Repair. Journal of Experimental Medicine, 2021. 218(9): p. e20200467.
[15] Thorp, E.B. and A. Karlstaedt, Intersection of Immunology and Metabolism in Myocardial Disease. Circulation Research, 2024. 134(12): p. 1824-1840.
[16] Ouyang, J., H. Wang, and J. Huang, The Role of Lactate in Cardiovascular Diseases. Cell Communication and Signaling, 2023. 21(1): p. 317.
[17] Cui, Y., et al., The Role of AMPK in Macrophage Metabolism, Function and Polarisation. Journal of Translational Medicine, 2023. 21(1): p. 892.
[18] Tang, B., et al., An Update on the Molecular Mechanism and Pharmacological Interventions for Ischemia-Reperfusion Injury by Regulating AMPK/mTOR Signaling Pathway in Autophagy. Cellular Signalling, 2023. 107: p. 110665.
[19] Liu, Z., et al., mTOR in the Mechanisms of Atherosclerosis and Cardiovascular Disease. Discovery Medicine, 2021. 31(164): p. 129-140.
[20] O'Neill, L.A. and D.G. Hardie, Metabolism of Inflammation Limited by AMPK and Pseudo-Starvation. Nature, 2013. 493(7432): p. 346-355.
[21] Wang, H., et al., Tetrahydropalmatine Promotes Macrophage Autophagy by Inhibiting the AMPK/mTOR Pathway to Attenuate Atherosclerosis. Histology and Histopathology, 2025. 40(5): p. 697-710.
[22] Owaki, R., et al., AMPK Activators Suppress Cholesterol Accumulation in Macrophages via Suppression of the mTOR Pathway. Experimental Cell Research, 2023. 432(1): p. 113784.
[23] Ren, P.H., et al., Yangxinkang Tablet Protects against Cardiac Dysfunction and Remodelling after Myocardial Infarction in Rats through Inhibition of AMPK/mTOR-Mediated Autophagy. Pharmaceutical Biology, 2020. 58(1): p. 321-327.
[24] Zhang, J., et al., SIRT1/SIRT3 Modulates Redox Homeostasis during Ischemia/Reperfusion in the Aging Heart. Antioxidants, 2020. 9(9): p. 858.
[25] Sanz, M.-N., et al., Inducible Cardiac-Specific Deletion of Sirt1 in Male Mice Reveals Progressive Cardiac Dysfunction and Sensitization of the Heart to Pressure Overload. International Journal of Molecular Sciences, 2019. 20(20): p. 5005.
[26] Lim, J.H., et al., Sirtuin 1 Modulates Cellular Responses to Hypoxia by Deacetylating Hypoxia-Inducible Factor 1alpha. Molecular Cell, 2010. 38(6): p. 864-878.
[27] Xu, C.-q., et al., Sirtuins in Macrophage Immune Metabolism: A Novel Target for Cardiovascular Disorders. International Journal of Biological Macromolecules, 2024. 256: p. 128270.
[28] Zhang, Z., et al., Mouse Macrophage Specific Knockout of SIRT1 Influences Macrophage Polarization and Promotes Angiotensin II-Induced Abdominal Aortic Aneurysm Formation. Journal of Genetics and Genomics, 2018. 45(1): p. 25-32.
[29] Lin, R., et al., Sirtuins Regulate Macrophage Polarization in Heart Failure: Metabolic Reprogramming, Epigenetic Regulation, and Immune Cell Interactions. Pharmacological Research, 2025. 220: p. 107936.

