Coronary heart disease (CHD) is driven by a chronic inflammatory process within atherosclerotic plaques, where immune cell function is fundamentally dictated by cellular metabolism. This review explores how metabolic reprogramming governs the fate and function of key immune cells in CHD.
We discuss the distinct metabolic pathways, from glycolysis to oxidative phosphorylation, that underpin the pro-inflammatory or pro-resolving phenotypes of macrophages and T cell subsets. The dynamic evolution of this immunometabolic landscape is examined across disease stages, from early activation to advanced plaque dysfunction. Finally, we highlight the therapeutic potential of targeting these pathways, using metformin as a prototype and exploring emerging strategies like itaconate modulation. Understanding these metabolic-immune interactions offers a crucial framework for developing precision therapies to resolve vascular inflammation in CHD.
Table of Contents
1. Macrophage metabolic reprogramming in coronary heart disease
2. Metabolic pathways driving immune activation in atherosclerosis
3. Metabolic control of regulatory T cells in coronary heart disease
4. Stage-dependent immune metabolic reprogramming in coronary heart disease
5. Distinct metabolic profiles of CD4+ and CD8+ T cells in coronary heart disease
6. Metformin and pharmacological modulation of immune metabolism in coronary heart disease
01 Macrophage metabolic reprogramming in coronary heart disease
The functional fate of macrophages within atherosclerotic plaques is fundamentally dictated by their metabolic reprogramming. In the coronary plaque microenvironment, these immune cells undergo dynamic shifts that steer phenotypic polarization and disease progression. Inflammatory, M1-like macrophages predominantly rely on aerobic glycolysis, a pathway often driven by factors like hypoxia-inducible factor 1α (HIF-1α) to fuel pro-inflammatory cytokine production, including IL-1β[1, 2]. Conversely, anti-inflammatory M2-like macrophages, associated with plaque stability and tissue repair, preferentially utilize oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO), a metabolic preference confirmed by oxidative phosphorylation assay[2, 3]. Key metabolic regulators such as mTORC1[4] and AMPK sense the plaque milieu, integrating cues from lipid overload and hypoxia to direct this metabolic switch. Notably, impaired mitochondrial OXPHOS, a hallmark of inflammatory macrophages that reinforces their pro-inflammatory state, can be directly measured using an OXPHOS assay[5]. This intrinsic link between metabolism and function makes macrophage metabolic pathways a compelling therapeutic target. Strategies aimed at restoring mitochondrial function or modulating specific metabolite axes, such as the immune responsive gene 1 (IRG1)-itaconate pathway, to reprogram macrophages toward a stabilizing phenotype represent a promising avenue for intervention in atherosclerosis[6].
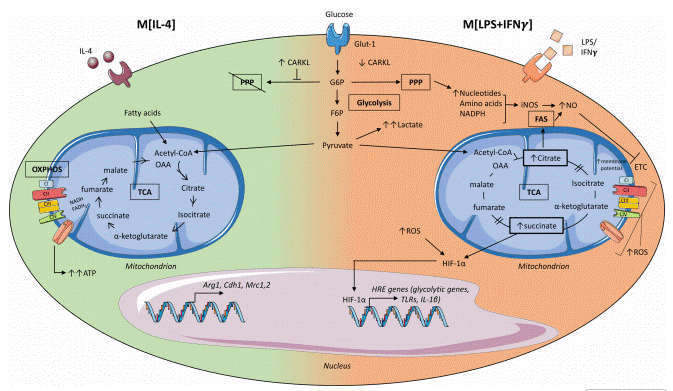
Fig. 1 Metabolic pathways controlling macrophage activation states[2]. Macrophage metabolism is stimulus-dependent. M [IL-4] cells use pyruvate in an intact TCA cycle for oxidative energy and tissue repair. M [LPS+IFN-γ] cells divert glycolysis to lactate and the PPP, with a broken TCA cycle. This leads to succinate and citrate accumulation, stabilizing HIF-1α to drive inflammatory gene expression and a pro-inflammatory state.
02 Metabolic pathways driving immune activation in atherosclerosis
The activation of immune cells, particularly T cell activation, within the atherosclerotic plaque is metabolically anchored by a rapid induction of glycolysis. This pathway fuels clonal expansion and cytokine production by providing ATP and biosynthetic precursors, driving the pro-inflammatory response[7]. This bioenergetic shift underpins the response of both innate and adaptive immune cells, including effector T cells, to the lesion's stressful milieu of hypoxia, oxidative stress, and lipid overload[8]. The glycolytic flux is often amplified by a broken tricarboxylic acid (TCA) cycle, where accumulated metabolites like succinate stabilize HIF-1α, creating a pro-inflammatory feed-forward loop that further sustains immune activation[9, 10]. Furthermore, the TCA cycle itself becomes a source of immunomodulatory signaling molecules, with citrate supporting itaconate and prostaglandin synthesis to modulate inflammatory outcomes[11]. Lipid metabolism exhibits a functional dichotomy: de novo fatty acid synthesis supports the anabolic needs of proliferating effector cells, such as during T cell activation, while FAO sustains the persistence and suppressive function of regulatory subsets[12]. Concurrently, amino acid metabolism, particularly involving glutamine and arginine, directs immune cell fate and fine-tunes activation states. Divergent arginine utilization, such as towards nitric oxide or polyamine synthesis, exemplifies how metabolic pathway choice directly instructs pro-inflammatory or reparative phenotypes in immune cells[13]. Thus, the atherosclerotic plaque acts as a metabolic arena where immune cell function, survival, and differentiation, including key processes like T cell activation, are shaped by flexible adaptation to and competition for nutrients. Targeting these pathways offers a promising strategy to modulate the inflammatory drive in atherosclerosis without causing broad immunosuppression[14].
%20and%20oxygen%20consumption%20rate(OCR)%20in%20T%20cells_.png)
Fig. 2 Measurement of extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) in T cells. OCR and ECAR were measured using specific assay kits. Activated T cells exhibited increased ECAR compared to naive T cells (The data are provided by Elabscience).
03 Metabolic control of regulatory T cells in coronary heart disease
In CHD, the atheroprotective function of regulatory T cells (Tregs) is critically determined by their metabolic fitness, namely the ability to maintain cellular energetics and function within a challenging microenvironment. Unlike pro-inflammatory effector T cells that switch to aerobic glycolysis upon activation, Tregs maintain immunosuppression and long-term homeostasis by preferentially utilizing OXPHOS fueled by FAO[15]. This metabolic profile is orchestrated by the master transcription factor Foxp3, which promotes an oxidative metabolic program while repressing glycolytic pathways[15]. However, this functional identity is vulnerable within the atherosclerotic plaque microenvironment. For instance, studies have shown that Tregs isolated from atherosclerotic plaques of mice and humans exhibit decreased mitochondrial membrane potential, elevated reactive oxygen species (ROS), and reduced Foxp3 expression compared to their counterparts in lymphoid organs, indicating profound metabolic distress. Metabolic stress from lipid overload, hypoxia, and competition for IL-2 can impair mitochondrial function, reduce the expression of key enzymes like CPT1A, and destabilize Foxp3 expression, leading to diminished suppressive capacity or even phenotypic conversion[15]. Key nutrient-sensing pathways integrate environmental signals to direct Treg metabolism and fate[16]. The mTOR kinase, particularly mTORC1, acts as a sensor that promotes glycolytic metabolism and effector cell differentiation; its inhibition favors Treg generation. Conversely, AMP-activated protein kinase (AMPK) serves as a crucial checkpoint that promotes oxidative metabolism and opposes mTOR signaling, thereby supporting Treg function[16]. Furthermore, cholesterol metabolism exerts a dual influence: while the mevalonate pathway supports Treg function, excessive intracellular cholesterol accumulation can be detrimental[17]. Therapeutic strategies aiming to bolster Treg-mediated immune tolerance in atherosclerosis are increasingly focused on correcting this metabolic imbalance. Pharmacological modulation of AMPK, mTOR, or specific lipid metabolic pathways represents a promising approach to enhance Treg metabolic fitness, stability, and function, offering a potential avenue to mitigate inflammation and promote plaque stability in CHD[16, 18].
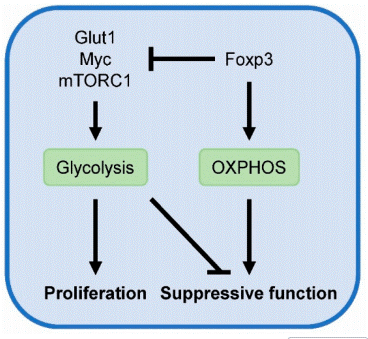
Fig. 3 Metabolic regulation of Treg activity[15]. Glycolysis drives the proliferation of Tregs but is linked to diminished suppressive activity. In contrast, Foxp3 represses glycolysis and enhances OXPHOS, which is essential for maintaining Treg-mediated suppression.
04 Stage-dependent immune metabolic reprogramming in coronary heart disease
Immune metabolic reprogramming in coronary heart disease is a dynamic and stage-specific process, fundamentally shaping disease progression from stable atherosclerosis to acute complications. This metabolic evolution directly mirrors the changing plaque microenvironment, which progresses from initial lipid accumulation to eventual hypoxia, necrosis, and inflammation. During early atherogenesis, the metabolic response is characterized by immune activation. Monocytes recruited to sites of endothelial dysfunction rapidly upregulate glycolytic metabolism upon engagement with atherogenic lipoproteins, supporting their differentiation and initial pro-inflammatory signaling[19]. Similarly, naive CD4+ T cells switch to aerobic glycolysis upon antigen encounter, facilitating clonal expansion and differentiation into effector subsets such as Th1 cells, thereby establishing early adaptive immune responses[20].
As the plaque matures, hypoxia within the growing necrotic core stabilizes HIF-1α. This factor orchestrates a pervasive metabolic shift, locking infiltrated macrophages and T cells into a sustained glycolytic state that reinforces a pro-inflammatory phenotype[2]. In this phase, metabolic programs diversify to support specialized immune functions; for example, Th17 cells utilize concurrent glycolysis and fatty acid synthesis to fuel interleukin-17 production[21].
In advanced plaques, metabolic reprogramming culminates in insufficiency and dysfunction. Macrophages experience severe mitochondrial distress, including impaired mitophagy and oxidative phosphorylation, which compromises essential homeostatic functions like efferocytosis[2]. This failure to clear apoptotic cells exacerbates the necrotic core and sustains inflammation. Concurrently, T cells, particularly CD8+ cytotoxic subsets, often exhibit features of exhaustion, characterized by suppressed glycolytic flux and fragmented mitochondria, leading to diminished effector capacity[22]. This progression from adaptive metabolic activation to maladaptive metabolic failure underscores the potential for therapeutic interventions precisely targeted to the metabolic state of immune cells at each distinct disease stage.
05 Distinct metabolic profiles of CD4+ and CD8+ T cells in coronary heart disease
In coronary heart disease, CD4+ and CD8+ T cells display divergent metabolic profiles that contribute to plaque pathology[23]. Studies utilizing tools such as CD4+ T cell isolation kits and CD8+ T cell isolation kits have helped delineate these subset-specific metabolic patterns. CD4+ T cells undergo distinct metabolic reprogramming: pro-inflammatory subsets like Th1 and Th17 cells primarily rely on glycolysis, fatty acid synthesis, and glutamine metabolism to support their effector functions[23, 24]. In contrast, atheroprotective subsets such as Tregs and Th2 cells depend more on oxidative phosphorylation (OXPHOS), fatty acid oxidation, and metabolism of arginine and kynurenine[24].
Although CD8+ T cells utilize glycolysis for activation, they often develop metabolic dysfunction in the atherosclerotic plaque microenvironment. This is characterized by impaired mitochondrial respiration and altered lipid metabolism, promoting a senescent or exhausted phenotype that may weaken immune surveillance and accelerate disease[25].
This subset-specific metabolic programming presents a promising therapeutic target[24]. Modulating these pathways, for instance by enhancing Treg oxidative metabolism or restoring CD8+ T cell metabolic fitness, could rebalance the immune response and provide new strategies for treating atherosclerosis[7].
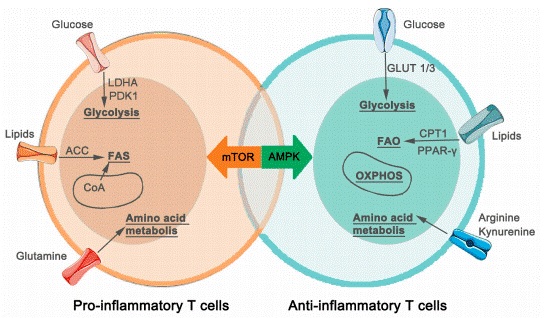
Fig. 4 Metabolic characteristics of CD4+ T-Cells in atherosclerosis[24]. In atherosclerosis, CD4+ T cells undergo distinct metabolic reprogramming. Pro-inflammatory subsets like Th1 and Th17 cells preferentially utilize glycolysis, fatty acid synthesis, and glutamine metabolism to fuel their effector functions. In contrast, anti-inflammatory, atheroprotective Tregs and Th2 cells rely more on OXPHOS, fatty acid oxidation, and metabolism of arginine and kynurenine to support their regulatory activity. Key molecules involved in these pathways are indicated in the diagram.
06 Metformin and pharmacological modulation of immune metabolism in coronary heart disease
Targeting immunometabolism offers a precise therapeutic strategy for coronary heart disease by reprogramming immune cell metabolism to resolve pathological inflammation without systemic immunosuppression[26]. Metformin exemplifies this approach, providing cardioprotection beyond glycemic control through AMPK activation and mTORC1 inhibition. This metabolic shift suppresses glycolysis in pro-inflammatory immune cells while promoting oxidative metabolism that favors anti-inflammatory phenotypes, including M2-like macrophages and regulatory T cells. In atherosclerotic models, metformin reduces plaque size, promotes an M2-dominant macrophage balance, and enhances Treg function[27]. Emerging strategies focus on specific metabolic nodes, including modulation of the IRG1-itaconate axis with compounds like 4-octyl itaconate, which attenuates plaque inflammation in preclinical studies[28]. The concept of targeting "trained immunity", a metabolically reprogrammed hyperinflammatory state, underscores the potential for durable interventions[29]. Resetting the metabolic-epigenetic axis represents a promising approach to durably mitigate vascular inflammation and alter atherosclerotic disease progression.
Elabscience® Quick Overview of Popular Products:
Table 1. Assay Kits for Coronary heart disease
|
Cat. No. |
Product Name |
|
E-BC-F069 |
Extracellular Acidification Rate (ECAR) Fluorometric Assay Kit |
|
E-BC-F070 |
Enhanced Oxygen Consumption Rate (OCR) Fluorometric Assay Kit |
|
E-BC-K853-M |
Glutamine (Gln) Colorimetric Assay Kit |
|
E-EL-H0149 |
Human IL-1β(Interleukin 1 Beta) ELISA Kit |
|
E-HSEL-R0002 |
High Sensitivity Rat IL-1β (Interleukin 1 Beta) ELISA Kit |
|
E-AB-F1351UD |
PE Anti-Mouse/Rat Foxp3 Antibody[FJK-16s] |
|
E-AB-F1238D |
PE Anti-Mouse Foxp3 Antibody[3G3] |
|
MIM007N |
EasySort™ Mouse Naïve CD4+T Cell Isolation Kit |
|
MIH007N |
EasySort™ Human Naïve CD4+T Cell Isolation Kit |
|
XJH002 |
Human Th17 Flow Cytometry Staining Kit |
|
E-BC-K902-M |
Succinic Acid Colorimetric Assay Kit |
|
E-BC-F065 |
Adipogenesis Fluorometric Assay Kit |
|
E-BC-K850-M |
L-Arginine (L-Arg) Colorimetric Assay Kit |
|
E-BC-F201 |
Enhanced ATP Chemiluminescence Assay Kit |
|
E-BC-F078 |
Mitochondrial Stress Fluorometric Assay Kit |
|
E-BC-F084 |
Glycolysis Stress Fluorometric Assay Kit |
|
E-BC-K221-M |
High-Density Lipoprotein Cholesterol (HDL-C) Colorimetric Assay Kit |
|
E-BC-K205-M |
Low-Density Lipoprotein Cholesterol (LDL-C) Colorimetric Assay Kit |
|
E-BC-K784-M |
Fatty Acid Oxidation (FAO) Colorimetric Assay Kit |
References:
[1] Wang, X., et al., Hypoxia Activates Macrophage-NLRP3 Inflammasome Promoting Atherosclerosis via PFKFB3-Driven Glycolysis. The FASEB Journal, 2024. 38(15): e23854.
[2] Koelwyn, G.J., et al., Regulation of Macrophage Immunometabolism in Atherosclerosis. Nature Immunology, 2018. 19(6): 526-537.
[3] Peng, X., et al., HMOX1-LDHB Interaction Promotes Ferroptosis by Inducing Mitochondrial Dysfunction in Foamy Macrophages During Advanced Atherosclerosis. Developmental Cell, 2025. 60(7): 1070-1086.e8.
[4] Zheng, H., et al., mTOR Signaling Promotes Foam Cell Formation and Inhibits Foam Cell Egress Through Suppressing the SIRT1 Signaling Pathway. Molecular Medicine Reports, 2017. 16(3): 3315-3323.
[5] Vendrov, A.E., et al., Mitochondrial Dysfunction and Metabolic Reprogramming Induce Macrophage Pro-Inflammatory Phenotype Switch and Atherosclerosis Progression in Aging. Frontiers in Immunology, 2024. 15.
[6] Cyr, Y., et al., The IRG1–Itaconate Axis Protects From Cholesterol-Induced Inflammation and Atherosclerosis. Proceedings of the National Academy of Sciences, 2024. 121(15): e2400675121.
[7] Hinkley, H., et al., T Cells in Atherosclerosis: Key Players in the Pathogenesis of Vascular Disease. Cells, 2023. 12(17): 2152.
[8] Gaddis, D.E., et al., Atherosclerosis Impairs Naive CD4 T-Cell Responses via Disruption of Glycolysis. Arteriosclerosis, Thrombosis, and Vascular Biology, 2021. 41(9): 2387-2398.
[9] Liu, Y.-X., et al., Metabolic Reprogramming and Cell Interaction in Atherosclerosis: From Molecular Mechanisms to Therapeutic Strategies. Journal of Cardiovascular Development and Disease, 2025. 12(10): 384.
[10] Xu, J., et al., Succinate/IL-1β Signaling Axis Promotes the Inflammatory Progression of Endothelial and Exacerbates Atherosclerosis. Frontiers in Immunology, 2022. 13: 817572.
[11] Gao, H., et al., Reprogramming Immunity With Itaconate: Metabolic Mechanisms and Therapeutic Perspectives. Inflammation Research, 2025. 74(1): 128.
[12] Nomura, M., et al., Macrophage Fatty Acid Oxidation Inhibits Atherosclerosis Progression. Journal of Molecular and Cellular Cardiology, 2019. 127: 270-276.
[13] Mills, C.D., Macrophage Arginine Metabolism to Ornithine/Urea or Nitric Oxide/Citrulline: A Life or Death Issue. Critical Reviews in Immunology, 2001. 21(5): 399-425.
[14] Julve, J., et al., Targeting Atherosclerosis: Cholesterol-Lowering Therapies With a New Immunometabolic Dress for an Old Disease. Journal of Clinical Medicine, 2025. 14(24): 8777.
[15] Baardman, J. and E. Lutgens, Regulatory T Cell Metabolism in Atherosclerosis. Metabolites, 2020. 10(7): 279.
[16] Al-Kuraishy, H.M., et al., Targeting of AMPK/MTOR Signaling in the Management of Atherosclerosis: Outmost Leveraging. International Journal of Biological Macromolecules, 2025. 309(Pt 2): 142933.
[17] Cai, F., S. Jin, and G. Chen, The Effect of Lipid Metabolism on CD4(+) T Cells. Mediators of Inflammation, 2021. 2021: 6634532.
[18] Foks, A.C., A.H. Lichtman, and J. Kuiper, Treating Atherosclerosis With Regulatory T Cells. Arteriosclerosis, Thrombosis, and Vascular Biology, 2015. 35(2): 280-7.
[19] Bekkering, S., et al., Innate Immune Cell Activation and Epigenetic Remodeling in Symptomatic and Asymptomatic Atherosclerosis in Humans in Vivo. Atherosclerosis, 2016. 254: 228-236.
[20] Tabas, I. and A.H. Lichtman, Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity, 2017. 47(4): 621-634.
[21] Zhang, S., et al., The Alterations in and the Role of the Th17/Treg Balance in Metabolic Diseases. Frontiers in Immunology, 2021. 12.
[22] Richter, F.C., et al., Take My Breath Away—Mitochondrial Dysfunction Drives CD8+ T Cell Exhaustion. Genes & Immunity, 2024. 25(1): 4-6.
[23] Yang, J., et al., Complex Interplay Between Metabolism and CD4+ T-Cell Activation, Differentiation, and Function: A Novel Perspective for Atherosclerosis Immunotherapy. Cardiovascular Drugs and Therapy, 2024. 38(5): 1033-1046.
[24] Chang, S., Z. Wang, and T. An, T-Cell Metabolic Reprogramming in Atherosclerosis. Biomedicines, 2024. 12(8): 1844.
[25] Molnár, A.Á., et al., Cells in Atherosclerosis: Focus on Cellular Senescence From Basic Science to Clinical Practice. International Journal of Molecular Sciences, 2023. 24(24): 17129.
[26] Hou, P., et al., Macrophage Polarization and Metabolism in Atherosclerosis. Cell Death & Disease, 2023. 14(10): 691.
[27] Luo, F., et al., Metformin in Patients With and Without Diabetes: A Paradigm Shift in Cardiovascular Disease Management. Cardiovascular Diabetology, 2019. 18(1): 54.
[28] Shan, W., et al., Itaconate as a Key Player in Cardiovascular Immunometabolism. Free Radical Biology and Medicine, 2024. 219: 64-75.
[29] DeBerge, M., et al., Immunometabolism at the Heart of Cardiovascular Disease. JACC: Basic to Translational Science, 2023. 8(7): 884-904.

