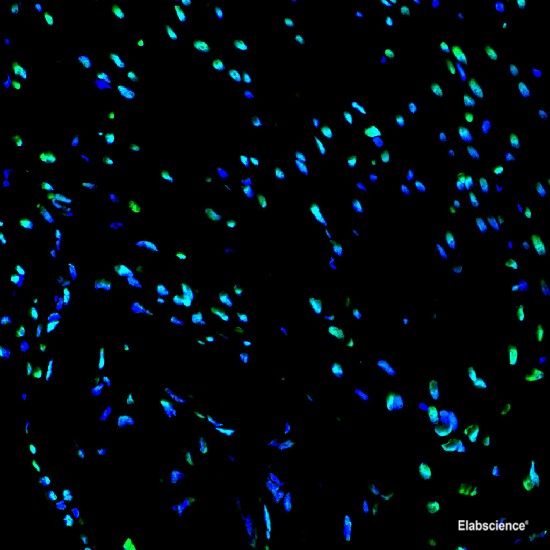
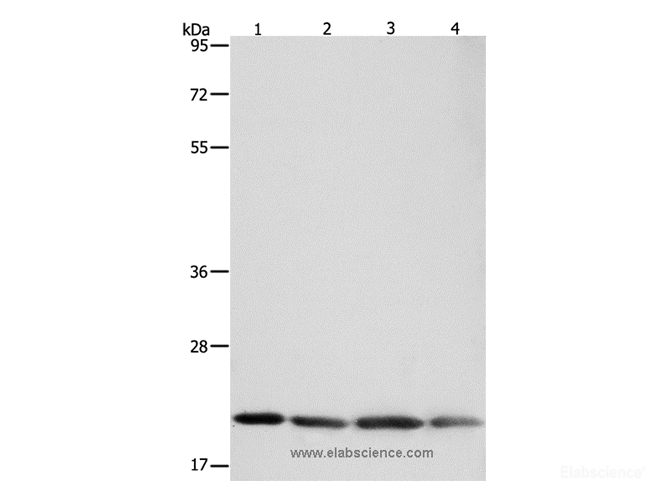
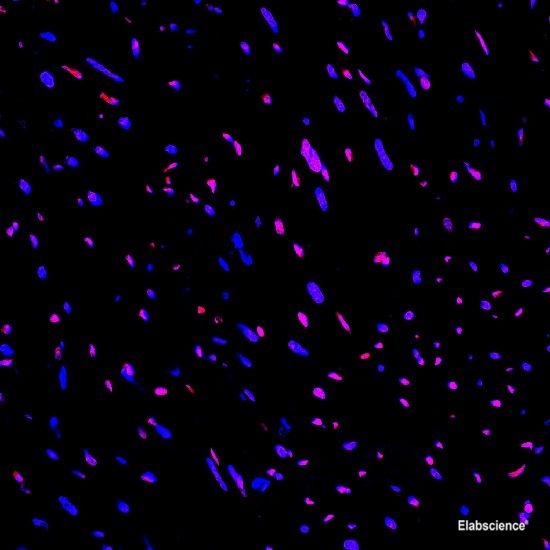
Background
PAH (phenylalanine hydroxylase), also known as PH, belongs to the biopterin-dependent aromatic amino acid hydroxylase family. It contains 1 ACT domain, N-terminal region of PAH is thought to contain allosteric binding sites for phenylalanine and to constitute an "inhibitory" domain that regulates the activity of a catalytic domain in the C-terminal portion of the molecule. In humans, PAH is expressed both in the liver and the kidney, and there is some indication that it may be differentially regulated in these tissues. PAH catalyzes the hydroxylation of the aromatic side-chain of phenylalanine to generate tyrosine. It is one of three members of the pterin-dependent amino acid hydroxylases, a class of monooxygenase that uses tetrahydrobiopterin and a non-heme iron for catalysis. Defects in PAH are the cause of phenylketonuria (PKU). PKU is an autosomal recessive inborn error of phenylalanine metabolism, due to severe phenylalanine hydroxylase deficiency. It is characterized by blood concentrations of phenylalanine persistently above 1200 mumol.